


Linezolid es un antibiótico sintético de un nuevo grupo, las oxazolidinonas, con espectro de actividad para grampositivos. Está indicado en infecciones de piel y tejidos blandos e infecciones nosocomiales adquiridas en la comunidad así como infecciones causadas por Staphylococcus aureus y Enterococus meticilin y vancomicin-resistentes, respectivamente. El objetivo de este trabajo es desarrollar y evaluar un método de cromatografía líquida de alta resolución (HPLC) para la monitorización de niveles plasmáticos de linezolid en muestras de pacientes.
Material y métodosSe utilizó como fase móvil una mezcla de acetonitrilo y agua con flujo isocrático de 1mL y una columna corta C18. La detección se realizó en un detector ultravioleta/visible a 254nm. El tratamiento de la muestra fue por precipitación de proteínas con ácido tricloroacético y posterior inyección del sobrenadante.
ResultadosEl método fue lineal y validado para un intervalo de 0,25 a 20mg/L. La precisión intra e interensayo (CV) fue inferior al 1% y 1,6%, respectivamente. La exactitud osciló entre -4,3% y 0,4%. La recuperación media fue superior al 82%. No se encontraron interferencias con otros fármacos habitualmente utilizados en la terapia combinada con linezolid. Tampoco se detectaron interferencias endógenas de la propia matriz biológica.
ConclusionesEl método descrito para cuantificar linezolid en muestras de plasma es sensible, reproducible, específico, rápido y requiere poca muestra, por lo que le hace adecuado para la monitorización terapéutica.
Linezolid is a synthetic antibiotic of the group of the oxazolidinones with Gram positive spectrum of activity. It is indicated in skin and soft tissue infections, community-acquired nosocomial infections and infections caused by methicillin-resistant and vancomycin-resistant Staphylococcus aureus and Enterococcus, respectively. The aim of this work is to develop and evaluate a high pressure (HPLC) method for the monitoring of plasma levels of linezolid in patients samples.
Materials and methodsThe mobile phase consisted of a mixture of acetonitrile and water with a flow rate of 1mL/min and a short C18 column. An ultraviolet/visible detector at 254nm was used to detect the peaks. Sample treatment consisted of precipitation of plasma proteins with trichloroacetic acid and then injection of the supernatant.
ResultsThe method was linear and validated from 0,25 to 20mg/L. The within-day and between-day coefficient of variation (CV) was less 1% and 1,6%, respectively. The accuracy varied between −4,3% and 0,4%. The average recovery was greater than 82%. No interferences were found with other drugs habitually used in the therapy combined with linezolid. Endogenous interferences of the biological matrix were not detected.
ConclusionsThe method described to quantify linezolid in plasma samples is sensitive, reproducible, specific, rapid and needs very little sample, which makes it suitable for therapeutic drug monitoring.
El linezolid (LNZ) es un antibiótico de una nueva clase, las oxazolidinonas, que actúa inhibiendo la iniciación de la síntesis de proteínas bacterianas. Presenta un espectro de gran actividad frente a microorganismos grampositivos y se utiliza sobre todo en el tratamiento de neumonías nosocomiales adquiridas en la comunidad, infecciones de piel y tejidos blandos de difícil accesibilidad e infecciones causadas por Staphylococcus aureus meticilin-resistentes y Enterococcus vancomicin-resistentes1,2.
La farmacocinética de LNZ destaca por presentar una biodisponibilidad oral del 100%, una semivida de eliminación de 5-6 horas, una baja unión a proteínas plasmáticas del orden del 31% y un volumen de distribución elevado (40-50 L), lo que indica una buena distribución en tejidos, alcanzando concentraciones altas en piel, líquido sinovial, humor acuoso, líquido cefalorraquídeo y parénquima pulmonar3. Debido a estas características, el LNZ es una buena alternativa terapéutica en infecciones de grampositivos que necesitan una buena penetrabilidad en tejidos. En estos casos es crucial conocer el grado de penetración del antibiótico para asegurar que se alcanzan concentraciones terapéuticas que permitan su empleo en sospecha o confirmación microbiológica de la infección4.
Farmacodinámicamente, LNZ presenta actividad antibacteriana tiempo-dependiente, siendo el ratio área bajo la curva (concentración-tiempo del fármaco)/concentración inhibitoria mínima, el mejor parámetro que determina la eficacia del LNZ5. Sin embargo, este parámetro es difícil de estimar en la práctica clínica y se han propuesto otros parámetros más simples como la determinación de las concentraciones mínimas antes de la dosis6, aunque se necesitarían más estudios que confirmen la correlación de éstas con la respuesta clínica y poder establecer un intervalo terapéutico para el LNZ.
Por otro lado, episodios de efectos adversos como pancitopenia inducida por LNZ y disfunción hepática han sido asociados a un incremento de las concentraciones valle de linezolid (15–20mg/L) comparado con los valores medidos en pacientes que han tolerado bien el tratamiento (3,8±2,5mg/L)7.
Se han descrito diferentes métodos por cromatografía líquida de alta resolución (HPLC) para cuantificar concentraciones de linezolid en plasma8–21. Algunos de estos métodos han utilizado gradiente de elución de fase móvil, otros utilizan procedimientos de tratamiento de muestra en extracción en fase sólida y el uso de estándar interno.
El objetivo principal de este estudio es el desarrollo, puesta a punto y validación de un método analítico rápido, preciso y sencillo para la determinación de LNZ en plasma mediante HPLC con aplicación a la monitorización terapéutica en pacientes que reciben tratamiento con este fármaco.
Material y métodosReactivosLinezolid como base libre ((S)-N-({3-[3-fluoro-4-(morpholin-4-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl}methyl) acetamide) fue proporcionado por Pfizer Inc. (Groton, CT, EE.UU.). Acetonitrilo de grado HPLC, metanol grado HPLC y ácido tricloroacético grado analítico mínimo 99% se obtuvieron de Sigma-Aldrich (Steinheim, Alemania); agua ultrapura fue obtenida a través de un sistema de purificación Milli-Q.
Condiciones cromatográficasEl sistema cromatográfico consiste en una bomba cuaternaria con autoinyector refrigerado (modelo Waters 2695), un detector de absorbancia ultravioleta/visible (modelo Waters 2487) y un sistema de adquisición de datos con integrador EmpowerTM software versión 3.0 (Waters, Milford MA, EE.UU.).
La separación fue realizada a 25°C utilizando una columna Symmetry® C18 de 3,5μm de diámetro de partícula, 7,5cm de longitud y 0,46cm de diámetro interno (Waters, Irlanda). La fase móvil fue preparada con agua (77%) y acetonitrilo (23%) desgasificada y filtrada con una membrana de nylon de 0,45μm de tamaño de poro y 47mm de diámetro (Millipore, EE.UU.). El flujo fue isocrático a 1mL/min. LNZ fue detectado en un detector ultravioleta/visible a 254nm de longitud de onda.
Preparación de calibradores y muestras controlSe prepararon dos soluciones patrón independientes de LNZ a concentración de 1g/L en metanol: una para preparar los calibradores y la otra para preparar las muestras control. Tanto para calibradores como muestras control se preparó una solución intermedia en metanol/agua (1:1, v/v) a una concentración final de 100mg/L. Las muestras de calibradores fueron obtenidas a partir de plasma libre de fármaco con diluciones apropiadas de la solución intermedia de trabajo de concentración 100mg/L para obtener calibradores con concentraciones de 0,25, 0,5, 1, 2,5, 5, 10 y 20mg/L. Las muestras control fueron preparadas de la misma forma a las concentraciones de 0,75, 7,5 y 15mg/L. Todas las soluciones, calibradores y controles fueron almacenados y protegidos de la luz a −80°C hasta su uso.
Preparación de la muestraLa preparación de la muestra consiste en la precipitación de proteínas con un ácido. En un eppendorf con 200μL de plasma (muestras de pacientes, calibradores y controles) se le añade 20μL de ácido tricloroacético al 50% en agua. Se agita en un vórtex durante 1 minuto y se centrifuga a 10.900g a temperatura ambiente durante 10 minutos. El sobrenadante se traspasa al vial de cromatografía y se inyecta 50μL en el sistema cromatográfico.
Validación del métodoLa validación del método se realizó de acuerdo a las normas de la FDA para ensayos bioanalíticos: Food and Drug Administration guidelines for validation of bioanalytical assays22.
LinealidadSe procesaron 5 curvas completas con los estándares plasmáticos (0,25, 0,5, 1, 2,5, 5, 10 y 20mg/L) que fueron analizadas en días diferentes. El cálculo del coeficiente de correlación para cada curva (r2) se hizo mediante regresión lineal por intersección en la recta de las áreas de los picos de LNZ con las concentraciones.
PrecisiónSe evaluaron tres niveles de concentración coincidentes con las muestras control. Se efectuaron 5 determinaciones analíticas de cada uno de los tres niveles, diariamente y durante cinco días consecutivos.
La precisión intradía (n=5) de los 3 controles analizados en el mismo día y de la interdía (n=15), analizados en los 5 días consecutivos, se expresó como coeficiente de variación (CV%) intra e interensayo:
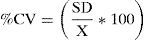
Siendo:
SD: desviación estándar de la concentración.
X: valor medio de las concentraciones analizadas (mg/L)
ExactitudSe evaluaron tres niveles de concentración coincidentes con las muestras control. Se efectuaron 5 determinaciones analíticas de cada uno de los tres niveles en un mismo día (exactitud intraensayo, n=5) y en un total de cinco días (exactitud interensayo, n=15).
La exactitud (% error relativo medio) intra e interensayo se calcularon en función de:

Siendo:Erm: exactitud o error relativo medioLNZ Experimental: concentración de linezolid obtenido (mg/L)LNZ Teórica: concentración de linezolid teórica (mg/L)
RecuperaciónEl cálculo de la recuperación de LNZ se realizó mediante la determinación del porcentaje del fármaco tras la extracción de muestras plasmáticas con concentraciones conocidas de LNZ frente a la inyección de la misma cantidad de fármaco en metanol/agua (1/1). Para ello, se inyectaron 5 extracciones de muestras correspondientes a los tres niveles de concentración coincidentes con las muestras control y muestras de patrones preparadas en metanol/agua equivalentes por concentración. Se evaluaron las áreas de los cromatogramas obtenidos:
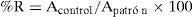
R=recuperación
Apatrón=área del pico cromatográfico de la solución patrón en metanol/agua
Acontrol=área del pico cromatográfico de la muestra control de calidad
Límite de cuantificación y límite de detecciónPara evaluar el límite de cuantificación (LC) y límite de detección (LD) se añadieron concentraciones crecientes de LNZ a un pool libre de fármaco y se midió la señal correspondiente. El LC se definió como el valor de concentración más bajo con una relación de desviación estándar del ± 20%. El LD se estableció en aquella concentración cuya proporción señal/ruido fuese de 3:1. La precisión y exactitud del LC y LD se evaluaron en siete muestras.
Especificidad y selectividadPara evaluar la especificidad se estudiaron seis blancos procedentes de matrices biológicas diferentes. Para ello, se procedió a identificar el LNZ mediante la comparación de una muestra del fármaco en disolución y establecer el tiempo de retención en el cromatograma. Se observaron los cromatogramas de los seis blancos (una vez procesada la muestra) para que no hubiera interferencias procedentes de la matriz biológica que pudieran interferir en la cuantificación del LNZ. Para evaluar la selectividad se procedió a comprobar posibles interferencias de la administración de diversos fármacos habituales concomitantes en la terapia con LNZ mediante la inyección de disoluciones en metanol/agua (1/1) de concentración conocida en el sistema cromatográfico: antirretrovirales (amprenavir, atazanavir, efavirenz, etravirina, didanosina, darunavir, indinavir, lopinavir, maraviroc, nelfinavir, nevirapina, ritonavir, raltegravir, saquinavir y tipranavir), inmunosupresores (rapamicina, ciclosporina, everolimus, tacrolimus y mofetilmicofenolato), antifúngicos (voriconazol, posaconazol, fluconazol), antivirales (ganciclovir), antibióticos (trimetoprim, sulfametoxazol), tuberculostáticos (rifampicina, isoniazida). Se descartan todas las sustancias que no aparezcan en el cromatograma en el tiempo de retención del LNZ.
EstabilidadLa estabilidad del LNZ se estudió en la matriz biológica durante tres ciclos de congelación y descongelación en los tres niveles de concentraciones correspondientes a las muestras control (0,75, 7,5 y 15mg/L) y por triplicado. Para el estudio de la estabilidad en el carro de muestras, se realizó un primer análisis tras la preparación de éstas y un segundo análisis transcurridos las 24 horas y a temperatura ambiente con los tres niveles de concentración de las muestras control y con tres determinaciones en cada caso. La pérdida de estabilidad se estableció en un aumento o disminución del 10% de la concentración observada inicialmente.
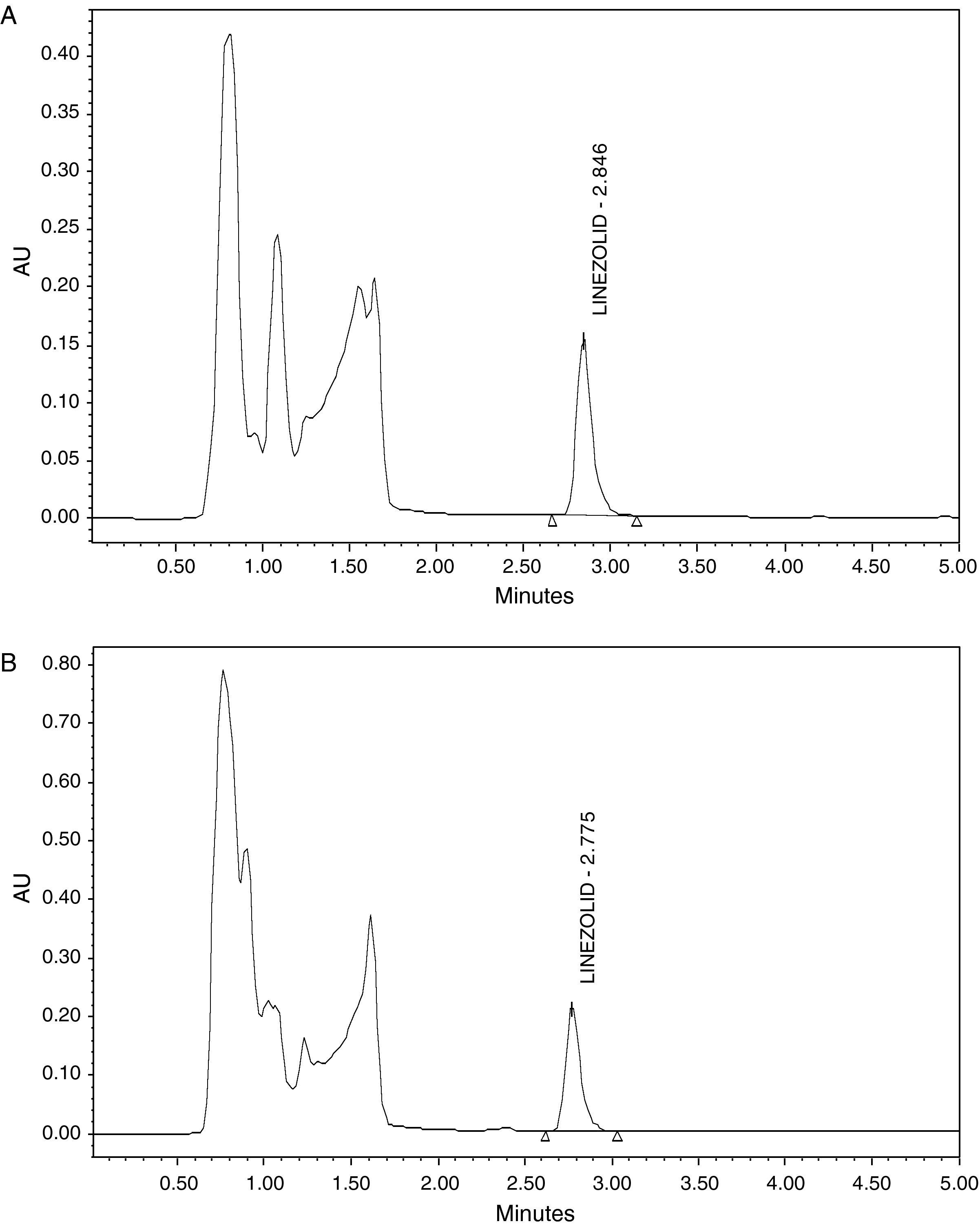
ResultadosEl tiempo de retención para el LNZ fue de 3 minutos aproximadamente en un tiempo de cromatograma total de 5 minutos (fig. 1). La linealidad de la curva de calibración ha sido verificada de 0,25 a 20mg/L en plasma (y=1,3533 e5 x−0,0052; r2=0,9999)
 y una muestra de un paciente con LNZ a la concentración de 10,8mg/L (B).")
El LD establecido fue de 0,1mg/L con una precisión de 1,2% y una exactitud (erm%) de 7%. El LC resultó de 0,25mg/L con una precisión (CV%) del 0,6% y una exactitud (erm%) de 4%.
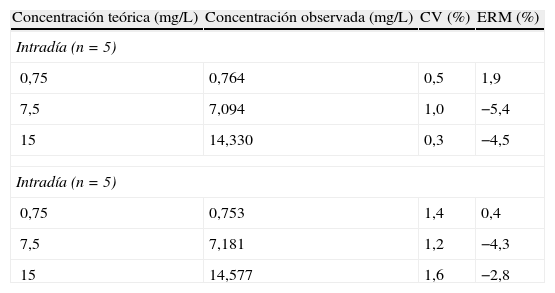
La precisión intraensayo (CV) osciló entre 0,3% (15mg/L) y 1% (7,5mg/L), mientras que la precisión interensayo osciló entre 1,2% (7,5mg/L) y 1,6% (15mg/L). La exactitud osciló entre -4,3% a 0,4% (tabla 1).
Precisión y exactitud
| Concentración teórica (mg/L) | Concentración observada (mg/L) | CV (%) | ERM (%) |
| Intradía (n=5) | |||
| 0,75 | 0,764 | 0,5 | 1,9 |
| 7,5 | 7,094 | 1,0 | −5,4 |
| 15 | 14,330 | 0,3 | −4,5 |
| Intradía (n=5) | |||
| 0,75 | 0,753 | 1,4 | 0,4 |
| 7,5 | 7,181 | 1,2 | −4,3 |
| 15 | 14,577 | 1,6 | −2,8 |
CV: coeficiente de variación; ERM: exactitud o error relativo medio.
La recuperación media del LNZ en plasma fue del 82,8%, 85,7% y 84,9% para los tres niveles de concentración estudiados de 0,75, 7,5 y 15mg/L, respectivamente.
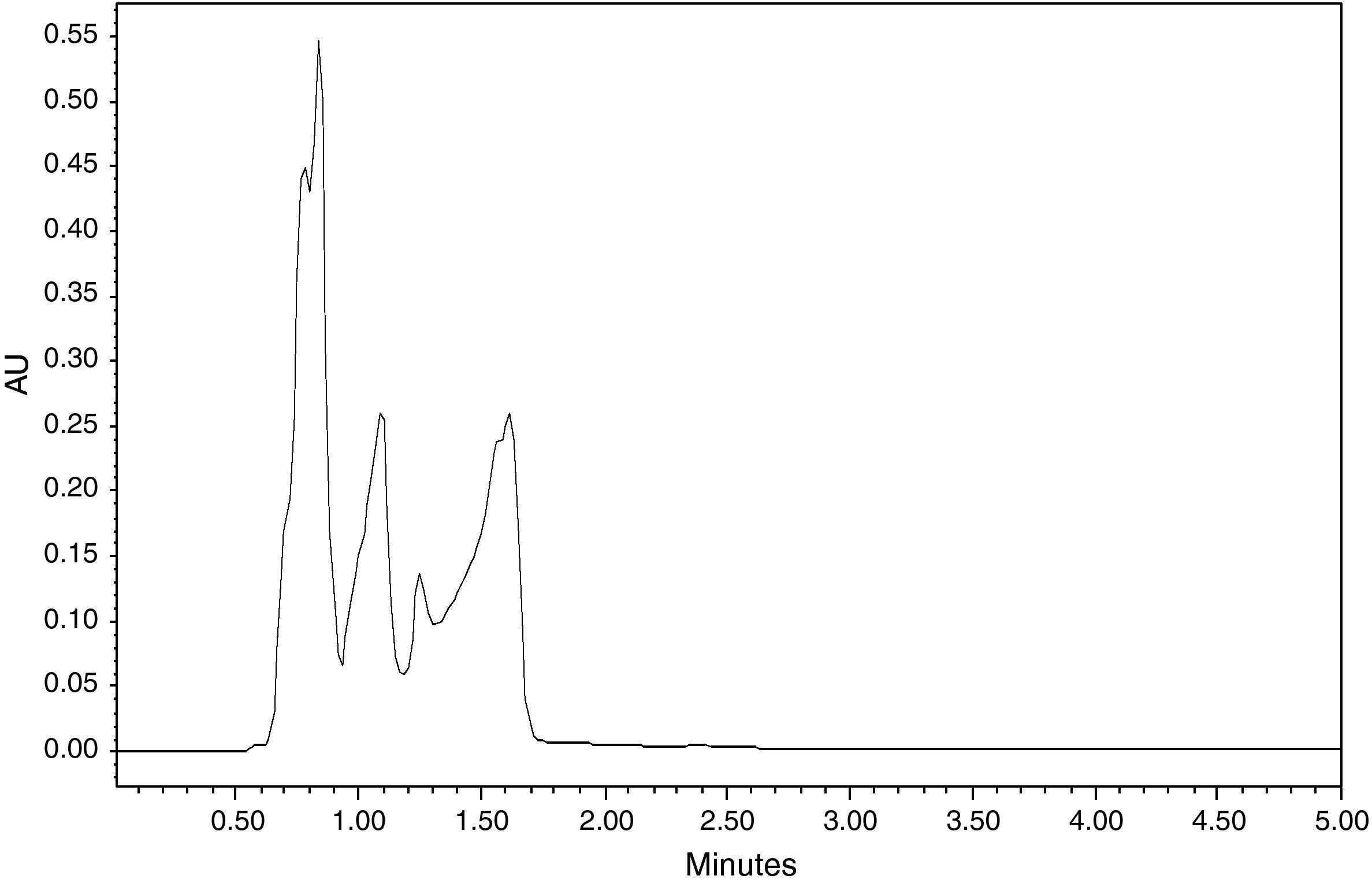
No se detectaron interferencias endógenas de la propia matriz biológica (fig. 2). Tampoco se encontraron interferencias con los fármacos analizados y utilizados habitualmente en la terapia combinada con LNZ.

El estudio de estabilidad de congelación y descongelación de las muestras de plasma con LNZ demostró que son estables 3 ciclos. Así mismo la muestra preparada fue estable al menos 24 horas a temperatura ambiente. En ninguno de los dos casos las concentraciones obtenidas fueron inferiores ni superiores al 10% de las concentraciones observadas inicialmente.
DiscusiónSe desarrolló un método analítico rápido, sensible y simple para determinar y cuantificar concentraciones de LNZ en plasma de manera fiable y precisa. El intervalo de linealidad de las curvas de calibración incluye un rango amplio de concentración de LNZ y la elección de los límites bajo y alto está basada en los resultados referenciados en la bibliografía de estudios clínicos farmacocinéticos, lo que evita tener que realizar diluciones en la mayoría de los casos. Los resultados de exactitud y precisión evidencian la reproducibilidad y fiabilidad de nuestro método.
La selectividad del método es muy alta, ya que no se han encontrado interferencias analíticas con otras drogas comúnmente utilizadas en pacientes que están en tratamiento con LNZ para infecciones por grampositivos. La especificidad también es muy correcta ya que con el tratamiento de precipitación de proteínas con tricloroacético y a la longitud de onda de detección del LNZ no se han encontrado interferencias de sustancias endógenas de la propia matriz biológica.
Nuestros resultados son similares a los métodos descritos en la literatura para la cuantificación de LNZ en plasma por HPLC con detección ultravioleta8–21.
El método descrito presenta diversas ventajas sobre los métodos previamente descritos por cromatografía líquida de alta resolución (HPLC) y detección ultravioleta/visible. Algunos utilizan una fase móvil compleja con mezcla de reactivos orgánicos para la separación cromatográfica18; por el contrario nuestro método utiliza una simple mezcla de agua con acetonitrilo. Para el tratamiento de muestra y eliminación de proteínas, en nuestro método realizamos una precipitación sencilla con ácido tricloroacético con lo que se acorta tiempo y se reducen costes, presentando una gran eficiencia, que le hace particularmente útil en estudios farmacocinéticos, mientras que otros métodos utilizan extracción de LNZ en plasma en columnas de fase sólida20,21, lo que supone un mayor consumo de tiempo y recursos económicos. Así mismo, nuestro método no requiere de la adición de patrón interno para la cuantificación de las muestras, a diferencia de otros autores9–15. La utilización de una columna corta (7,5cm) en nuestro método reduce el tiempo de cromatograma sin perder eficacia y resolución, en contraste con otros autores que utilizan columnas más largas (10-25cm) con el consiguiente aumento de tiempos de trabajo y costes8–21.
Como conclusión podemos decir que el método descrito para la cuantificación de LNZ en plasma por HPLC es sensible, reproducible, selectivo, eficiente, rápido y simple y de bajo coste económico y adecuado para su posible aplicación en la monitorización terapéutica de LNZ en pacientes tratados con este fármaco.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen al laboratorio farmacéutico que ha suministrado la sustancia pura de linezolid (Pfizer Inc. Groton, CT, EE.UU.).
Este trabajo corresponde a una comunicación científica presentada y premiada en el IV Congreso Nacional del Laboratorio Clínico, celebrado en Zaragoza del 20-22 de octubre de 2010.
Este trabajo corresponde a una comunicación científica presentada y premiada con accésit en el IV Congreso Nacional del Laboratorio Clínico celebrado en Zaragoza del 20 al 22 de octubre de 2010.
