


Las leucemias agudas (LA) constituyen un grupo heterogéneo de hemopatías con diferente etiología, patogenia, historia natural y pronóstico. Con su clasificación se ha intentado reducir dicha heterogeneidad e identificar subgrupos biológicamente diferentes y con distintas opciones terapéuticas, lo que ha permitido mejorar el pronóstico de los pacientes con esta enfermedad.
Actualmente la clasificación según el grupo cooperativo franco-americano-británico (FAB) se sigue utilizando, especialmente en el momento en el que se detectan los elementos blásticos en la sangre periférica del paciente, quien suele acudir a su médico de cabecera, o a un servicio de urgencias, por la aparición aguda de síntomas clínicos. La primera orientación diagnóstica de LA mieloide o linfoide se realiza en base a lo que se dispone en ese momento; es decir mediante la atenta observación morfológica de los elementos inmaduros. Posteriormente, la aplicación de todos los estudios de los que en la actualidad se disponen (citoquímica, inmunofenotipo, citogenética y biología molecular) permitirá clasificar el tipo de leucemia que presenta dicho paciente. La clasificación más reciente de la OMS es de gran utilidad para el manejo clínico de los pacientes.
Acute leukemias (AL) are hematological malignancies with different aetiologies, natural history and prognosis. The aim of the classification in AL is to reduce this heterogeneity and to identify biologically different groups in order to improve the therapeutical options and patient prognosis.
FAB classification is still used for the first morphological orientation of the blast cells at the time of the diagnosis. In a second step, it is necessary to apply all the additional studies, especially immunophenotype, cytogenetic and molecular analysis, in order to find out the type of acute leukemia in the WHO classification.
Las leucemias agudas (LA) son el resultado de una mutación somática en una única célula madre hematopoyética, que desencadena una proliferación clonal de células leucémicas inmaduras. La célula en la que se produce la transformación leucémica es un precursor que pierde la capacidad de seguir su proceso normal de maduración. Cuando este precursor es de origen mieloide, se desarrolla una leucemia aguda mieloide (LAM).
Las alteraciones genéticas que acompañan a la transformación leucémica de una célula suelen ser alteraciones cromosómicas adquiridas. En las LA las células blásticas proliferan en la médula ósea y reemplazan a la celularidad normal de la misma, lo que provoca una disminución de las 3 series hematopoyéticas en sangre periférica (anemia, neutropenia y trombocitopenia). En consecuencia, las LA suelen acompañarse de infección y/o hemorragia. La proliferación de células blásticas en otros órganos se traduce en la presencia de hepatomegalia y/o esplenomegalia o adenopatías.
En la década de los 80 los estudios morfológicos, citoquímicos, de citogenética convencional y de inmunofenotipo tuvieron un papel importante en la definición del tipo celular neoplásico y, por tanto, en el diagnóstico de las leucemias. Sin embargo, es en la década de los 90 cuando se consolida el papel fundamental del estudio morfológico y, al mismo tiempo, adquieren un gran protagonismo otros métodos, tales como la citometría de flujo, la hibridación in situ, la citogenética molecular y la biología molecular1–3.
En la última década comienzan a descifrarse algunos mecanismos que originan las distintas variedades de LAM. En las neoplasias, las alteraciones moleculares suelen afectar a genes claves implicados en el control del ciclo celular, o en los mecanismos de apoptosis. También pueden ser secundarias a la fusión de genes normales y a la formación de genes nuevos que dan lugar a proteínas oncogénicas. El resultado de las alteraciones moleculares es un desequilibrio del ciclo celular o la abolición de los mecanismos de apoptosis, que tiene como consecuencia la prolongación de la vida celular.
La clasificación de las LA más ampliamente utilizada ha sido la del Grupo Cooperativo Franco-Americano-Británico (FAB)4,5. Más recientemente se ha propuesto la clasificación de la Organización Mundial de la Salud (OMS 2001 y 2008)6–9 (fig. 1).

Ante la sospecha de una LAM se realizará un mielograma, un estudio citoquímico, un análisis inmunofenotípico y un análisis citogenético para obtener información pronóstica10,11.
El grupo cooperativo FAB diferenció las siguientes variedades morfológicas de LAM:
- 1.
LAM0 (mínimamente diferenciada).
- 2.
LAM1 (mieloblástica sin maduración).
- 3.
LAM2 (mieloblástica con maduración).
- 4.
LAM3 (promielocítica).
- 5.
LAM4 (mielomonocítica).
- 6.
LAM5 (monocítica).
- 7.
LAM6 (eritroide).
- 8.
LAM7 (megacarioblástica).
La LAM mínimamente diferenciada o LAM0 tiene una gran dificultad diagnóstica desde el punto de vista morfológico, dado que los blastos presentan rasgos morfológicos linfoides y mieloides12. Constituye únicamente el 5% de las LAM en el adulto y tiene un mal pronóstico. Miembros del grupo FAB, utilizando citoquímica ultraestructural y anticuerpos monoclonales, demostraron que algunos casos con una cifra inferior al 3% de blastos mieloperoxidasa (MPO) positivos clasificados como leucemias agudas linfoides (LAL) eran en realidad LAM con signos mínimos de maduración. En el estudio inmunofenotípico al menos un marcador mieloide (MPO citoplasmática, CD13 o CD33) es positivo en los blastos. Los marcadores linfoides son negativos (CD3, CD22, CD79a).
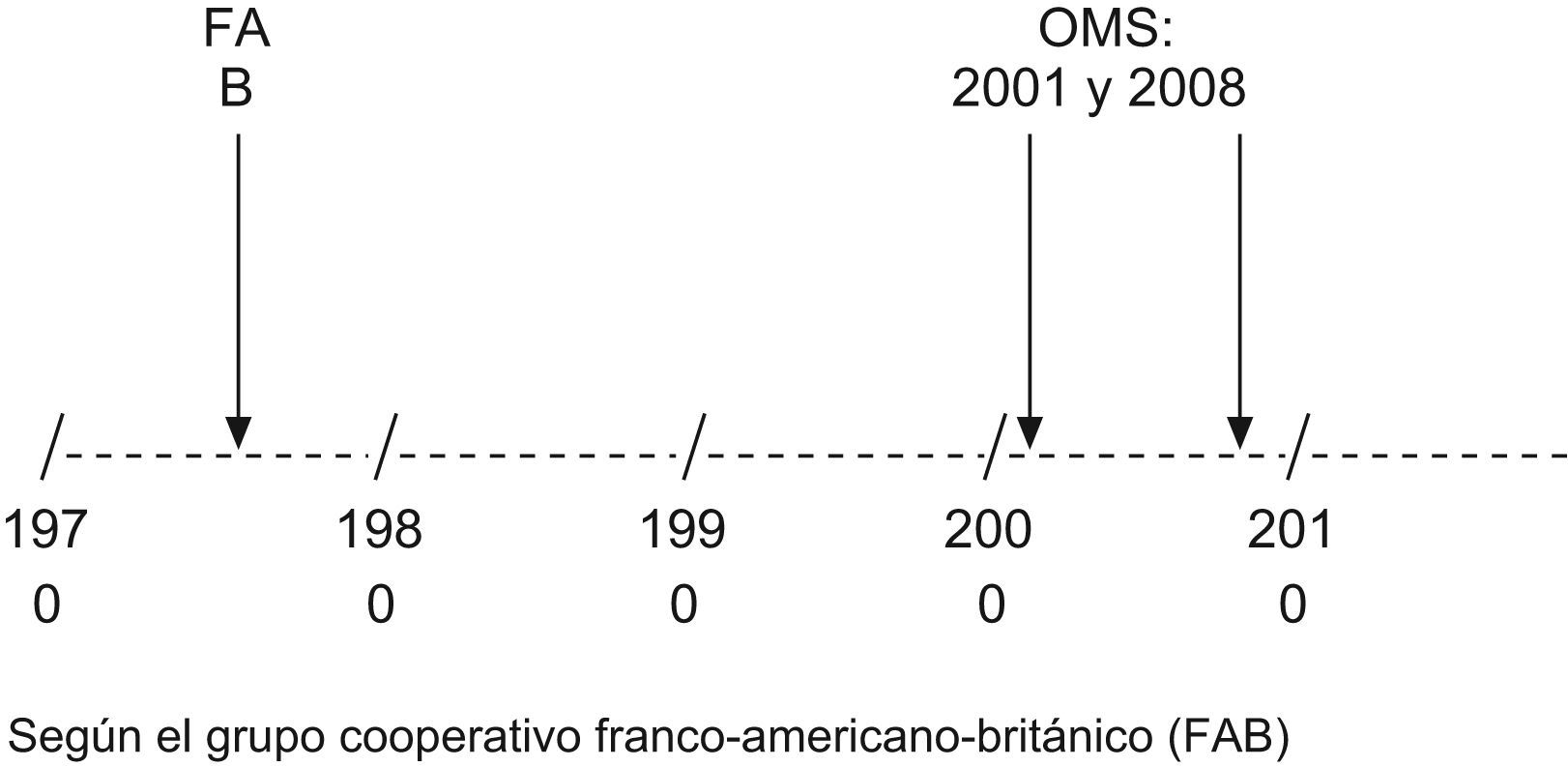
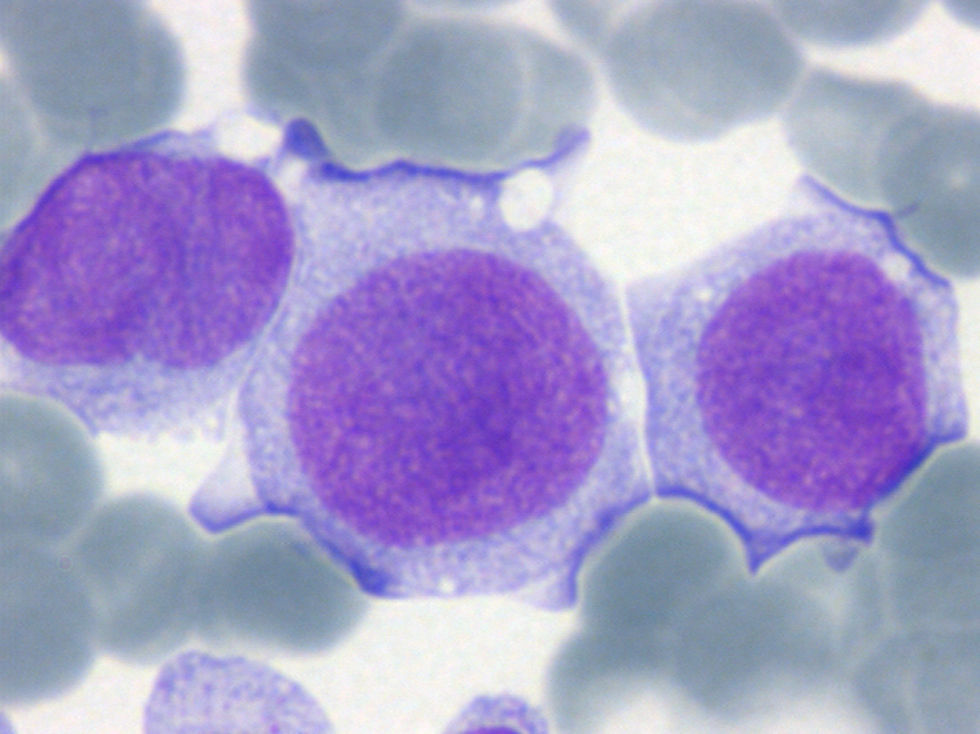
Leucemia aguda mieloide sin maduración (LAM1)En este subtipo suele observarse un monomorfismo celular, con presencia en sangre periférica (SP) de blastos mieloides (>3%) en ausencia de otras células en estadíos posteriores al mieloblasto. Los blastos son de tamaño mediano, con elevada relación núcleo-citoplasmática (N/C), contorno nuclear redondeado, núcleo de cromatina laxa e inmadura con presencia de uno o varios nucleolos prominentes (fig. 2). Los blastos pueden presentar una fina granulación azurófila, o algún bastón de Auer visible en el citoplasma, y una cifra superior al 3% de las células blásticas son MPO positivas. La fosfatasa ácida y la β-glucuronidasa muestran una positividad difusa. La reacción del ácido peryódico de Schiff (PAS) es positiva, débil y difusa. En el estudio inmunofenotípico se demuestra que los blastos expresan antígenos mieloides (CD33 y CD13) y pueden expresar también el antígeno CD34.
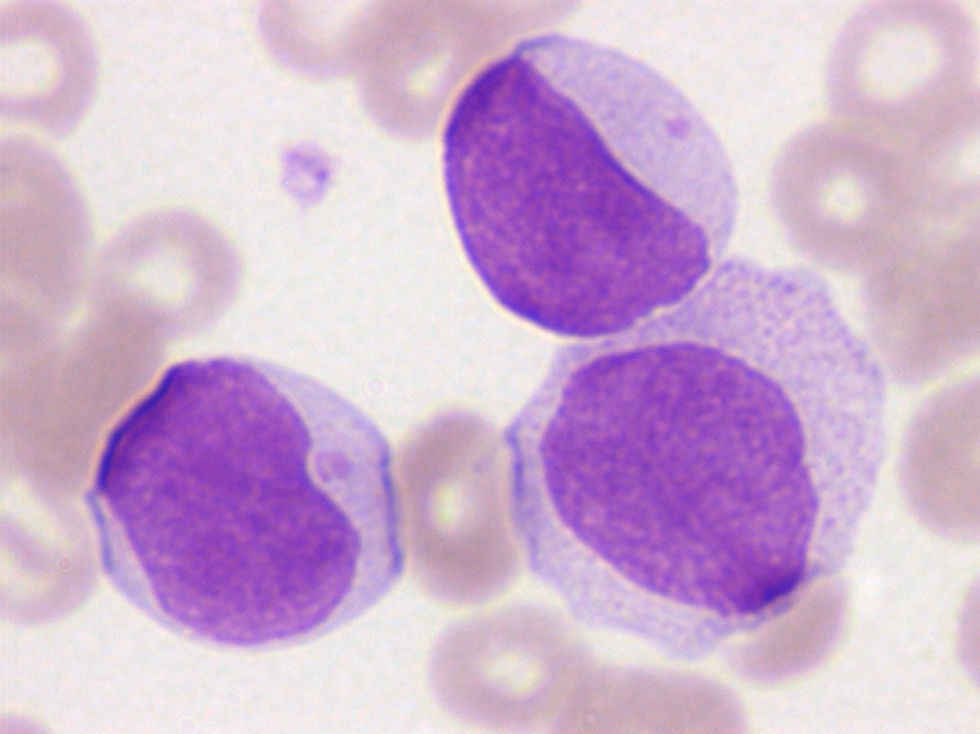
Leucemia aguda mieloide con maduración (LAM2) en sangre periférica: el citoplasma puede contener una fina granulacióna azurófila o algún bastón de Auer.")
Constituye alrededor del 30% de todos los casos de LAM y muestra células en estadios madurativos posteriores al mieloblasto (promielocitos, mielocitos y neutrófilos) en un porcentaje superior al 10%. El tamaño de los blastos en la LAM2 es de pequeño a mediano, con una elevada relación N/C y un perfil nuclear redondeado, que a veces adopta una posición cuadrangular respecto al citoplasma. El núcleo muestra una cromatina laxa e inmadura, con uno o varios nucleolos visibles. El citoplasma es basófilo y puede contener un esbozo de granulación primaria azurófila, u ocasionalmente algún bastón de Auer.
Los blastos de la LAM2 son positivos para la MPO y el Negro Sudán B, y expresan los antígenos CD34, HLA-DR, CD13 y CD15. Pueden expresar otros antígenos, tales como CD117, CD34 y HLA-DR. Una tercera parte de las LAM2 se asocian a t (8;21). Los casos de LAM2 con esta alteración citogenética presentan una supervivencia más prolongada y constituyen un subtipo de LAM con anomalías citogenéticas recurrentes según la clasificación de la OMS, tal como se describe más adelante. En este subtipo se observan blastos de tamaño pequeño junto a otros de mayor tamaño, núcleo de perfil más irregular y citoplasma moderadamente amplio, que puede contener una granulación muy marcada. Otras alteraciones son deleciones o translocaciones a nivel del cromosoma 12, y la t(6;9). Una asociación menos frecuente es la t(8;16)(p11;p13), caracterizada por la presencia de eritrofagocitosis, positividad para la MPO y esterasas inespecíficas, negatividad para los antígenos CD34 y CD117, con positividad para el CD56 y reordenamiento de los genes MOZ/CBP.
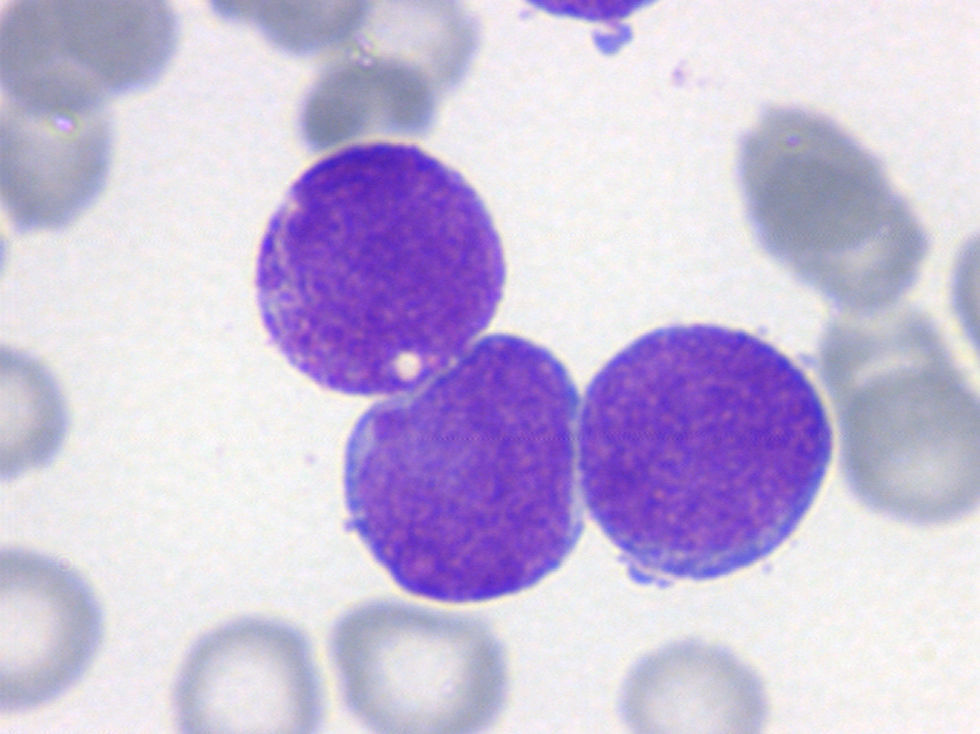
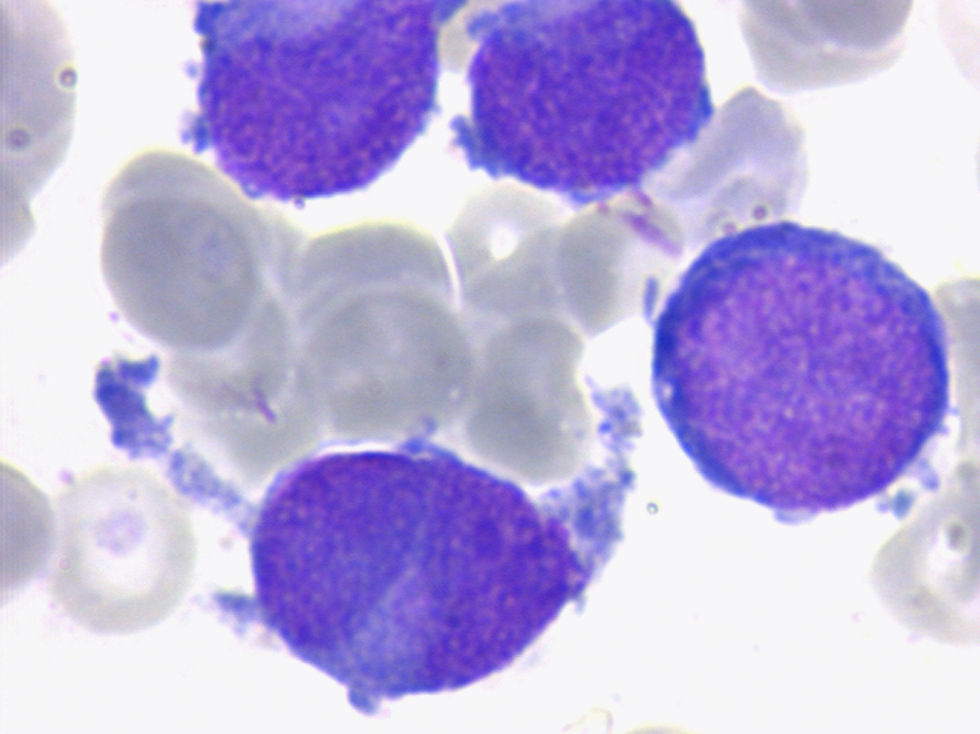
Leucemia aguda promielocítica (LAM3)Suele acompañarse de una cifra baja de leucocitos en SP, lo que dificulta su diagnóstico. Las células que proliferan muestran una morfología muy característica y se denominan promielocitos atípicos (hipergranulares). Puede cursar con accidentes hemorrágicos muy graves por coagulación intravascular diseminada. Los promielocitos atípicos presentan una granulación intensamente azurófila y muy abundante. El núcleo suele ser de aspecto monocitoide (reniforme) y con un perfil bilobulado (en hachazo) con la presencia de una hendidura amplia, o bien de perfil irregular (fig. 3). El citoplasma es poco basófilo debido al elevado contenido de granulación azurófila. Algunos de los promielocitos atípicos contienen además inclusiones citoplasmáticas cristalinas alargadas o astillas, específicas de este tipo de leucemia, que suelen disponerse en cúmulos y que difieren de los bastones de Auer por la detección de una subestructura tubular cuando se estudian mediante microscopía electrónica de transmisión.
 de cromatina laxa e inclusiones citoplasmáticas alargadas o astillas.")
Los promielocitos atípicos son muy positivos para la MPO y Negro Sudán B. La reacción del PAS muestra positividad difusa y las fosfatasas ácidas son intensamente positivas. Son HLA-DR y CD34 negativos y son positivos para los anticuerpos monoclonales CD13 y CD33.
La alteración citogenética característica de la LAM3 es la t(15;17), que provoca la fusión del oncogen PML(promyelocytic leukemic gen) con el gen del receptor del ácido retinoico (RARα), con el resultado de la formación del tránscrito PML-RARα. Para detectar la t(15;17) se utilizan técnicas citogenéticas, de hibridación in situ y de biología molecular. Estas últimas identifican específicamente el tránscrito PML-RARα en todos los casos de LAM3 que responden al tratamiento con ATRA (ácido trans-retinoico). El ATRA induce la maduración de las células leucémicas (a promielocitos, mielocitos y neutrófilos).
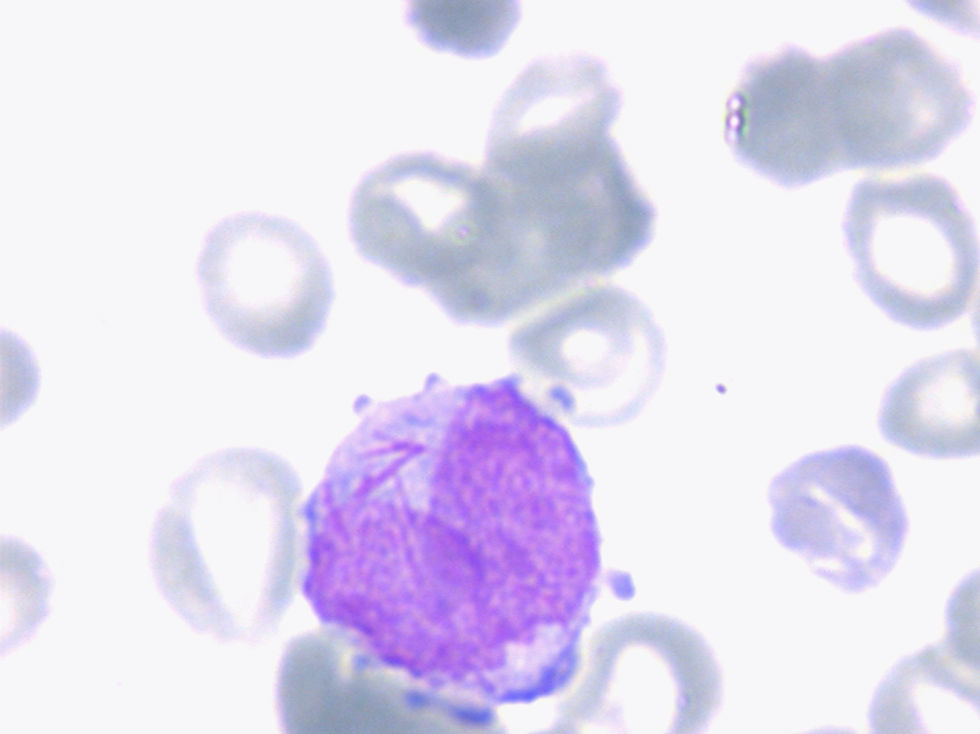
Variante microgranular de la LAM3Tiene un peor pronóstico que la LAM3 clásica y suele cursar con leucocitosis. Se caracteriza por la escasez de granulación en las células leucémicas. Constituye el 15–20% de la totalidad de las LAM3. El citoplasma es más basófilo que en la LAM3 clásica, debido a su menor contenido en granulación azurófila (fig. 4).
Leucemia aguda mielomonocítica (LAM4)
Tiene un componente granulocítico y otro monocítico, en proporciones variables y con diversos grados de maduración. Los blastos monocíticos son de gran tamaño, moderada relación N/C y basofilia variable. El núcleo puede ser redondeado, arriñonado o de forma irregular. Los nucleolos acostumbran a ser prominentes.
Los blastos mieloides son positivos para la cloroacetatoesterasa y los monocíticos para la naftol As-D-acetatoesterasa o la α-naftilbutiratoesterasa.
En la LAM4 los blastos son CD34 positivos y expresan marcadores mieloides (CD13, CD15 y CD33) y monocíticos (CD11b, CD11c, CD14, CD64 y CD4).
Leucemia aguda monocítica (LAM5)Constituye alrededor de un 15% del total de LAM. Las células leucémicas son de estirpe monocítica (monoblastos y promonocitos). La LAM5 incluye 2 subtipos:
- 1.
LAM5a o leucemia aguda monoblástica, en la que predominan los monoblastos.
- 2.
LAM5b o leucemia aguda monocítica, en la que junto a los monoblastos se observa una elevada proporción de promonocitos y monocitos.
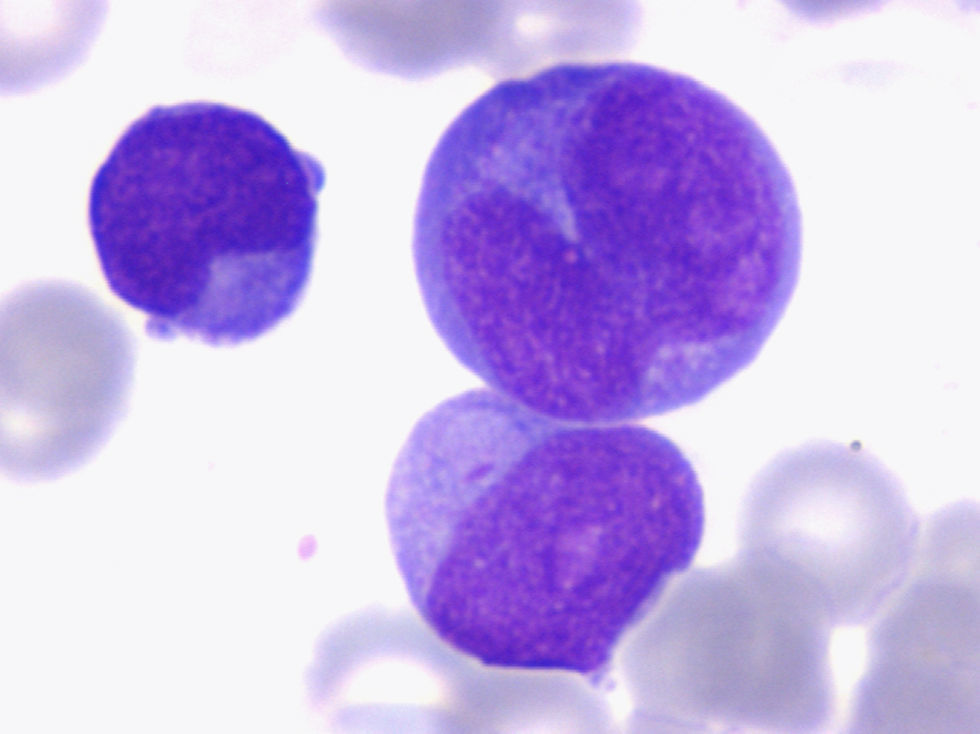
El subtipo LAM5a constituye alrededor de un 5–8% de las LAM. Los elementos blásticos son de gran tamaño, con un núcleo de perfil redondeado de cromatina laxa e inmadura (1–3 nucleolos), y un citoplasma moderadamente amplio e intensamente basófilo. En el citoplasma es posible observar algún bastón de Auer y/o prolongaciones o mamelones (fig. 5).

En la LAM5b (3–6% de las LAM) los promonocitos presentan un núcleo de perfil redondeado o arriñonado, y un citoplasma menos basófilo (fig. 6), con mayor contenido de granulación que los monoblastos y con la presencia de alguna vacuola. La observación de eritrofagocitosis junto a blastos monocíticos sugiere la existencia de una t (8;16).

Los blastos monocíticos son positivos para las esterasas inespecíficas, reacción que se inhibe en presencia de fluoruro sódico. La MPO puede ser negativa o débilmente positiva. Los blastos muestran además positividad difusa para las fosfatasas ácidas. La muramidasa sérica (y urinaria) está elevada. Los blastos de la LAM5 son HLA-DR positivos y expresan intensamente antígenos monocíticos (CD14, CD68, CD4, CD11c y CD64). La forma monoblástica suele asociarse a la t (9;11), la t(6;11) o la t(8;16) y también es frecuente el reordenamiento 11q23 (LAM con anomalías genéticas recurrentes según la OMS), la trisomía 8 y la monosomía 7.
Leucemias agudas eritroides (LAM6)Ha sido definida por la clasificación FAB como una proliferación de elementos eritroides displásicos, junto a una proliferación de elementos blásticos de origen mieloide. Se ha categorizado en dos subtipos:
- 1.
La eritroleucemia (LAM6a), con una proliferación blástica mixta mieloide y eritroide.
- 2.
La LAM6 variante o leucemia eritroide pura (LAM6b) según la clasificación de la OMS.
La eritroleucemia o LAM6a muestra una proliferación leucémica mixta de las series granulocítica y eritroblástica13. Constituye únicamente un 5–6% del total de casos de LAM, y puede ser secundaria a un síndrome mielodisplásico previo. Para su diagnóstico se requiere que, en médula ósea, los precursores eritroides sean de un 50% o más de la totalidad celular y los mieloblastos un 30% de la celularidad no eritroide (20% según la clasificación de la OMS).
En la eritroleucemia la morfología eritrocitaria de sangre periférica está muy alterada, con presencia de esquistocitos, «hematíes pinzados» o en forma de seta, hematíes espiculados del tipo equinocitos y acantocitos14.
LAM6 variante (leucemia eritroide pura según la clasificación de la OMS)En la LAM6 variante, más de un 80% de la celularidad de médula ósea está constituida por elementos eritroides, siendo el componente mieloide inferior al 3%. La leucemia eritroide pura se asocia a alteraciones importantes de la morfología eritrocitaria en SP, tales como macrocitosis, punteado basófilo, cuerpos de Howell-Jolly o anillos de Cabot. En la eritroleucemia o LAM6a los blastos presentan antígenos mieloides (CD13, CD33, CD15) y, en la leucemia eritroide pura o LAM6b expresan antígenos específicos de la serie eritroide (Glicoforina A o Glicoforina C), y son negativos para los antígenos mieloides. Las anomalías cromosómicas se sitúan frecuentemente en los cromosomas 5 y 7.
Leucemia aguda megacarioblástica (LAM7)Representa un 3–5% de las LAM. Los blastos muestran un aspecto morfológico muy inmaduro, y son muy polimórficos. El núcleo es excéntrico, de cromatina laxa y reticulada y con 1–3 nucleolos prominentes. El citoplasma es basófilo, agranular y muestra un aspecto muy similar a las plaquetas circulantes con presencia de mamelones o seudópodos (fig. 7). Se observan micromegacariocitos y fragmentos megacarioblásticos en SP, así como una gran dismorfia plaquetaria (plaquetas gigantes y algunas con marcada desgranulación).

Los blastos muestran positividad para CD61 (glicoproteína IIIa), CD41 (glicoproteína IIb/IIIa) y CD42 (glicoproteína Ib). La t(1;22)(p13;q13) es frecuente en niños menores de 1 año.
Según la organización mundial de la salud (WHO 2008)La clasificación de la OMS introduce algunos cambios que tienen en cuenta nuevos conceptos en la biología de las LAM15, especialmente a nivel citogenético, inmunológico y molecular.
- I)
LAM con anomalías genéticas recurrentes:
- a)
t(8;21), t(15;17), inv (16), t(9;11), t(6;9), inv(3), t(1;22).
- b)
mutaciones NPM1, FLT3, CEBPA.
- a)
- II)
LAM con DISPLASIA.
- III)
LAM relacionadas con tratamientos previos (agentes alquilantes o inhibidores de la topoisomerasa II).
- IV)
LAM no categorizadas previamente, que incluyen:
- a)
Subtipos de la clasificación FAB.
- b)
Leucemia aguda basofílica.
- c)
Panmielosis aguda con mielofibfrosis.
- a)
- V)
Sarcoma mieloide.
- VI)
Proliferaciones mieloides en relación con el síndrome de Down.
- VII)
Neoplasias de células blásticas dendríticas plasmocitoides.
En este grupo de LAM tienen lugar determinadas anomalías citogenéticas que corresponden a translocaciones recíprocas, tales como t(8;21), inv(16) o t(16;16), t(15;17), t(9;11), t(6;9), t(3;3) o inv(3) y t(1;22). Estas anomalías citogenéticas son responsables de la formación de genes de fusión (FLT3, NPM1, CEBPA), que codifican la síntesis de proteínas quiméricas. Las translocaciones mencionadas se ponen de manifiesto mediante la reacción en cadena de la transcriptasa-polimerasa, con mayor sensibilidad que la citogenética convencional.
Leucemia aguda mieloide con t(8;21)Constituye alrededor del 5% de las LAM. La t (8;21) (q22;q22), junto al gen de fusión RUNX1-RUNX1T1, se observa en un 10% de las LAM con maduración (categoría M2 según la clasificación FAB). Es más frecuente en pacientes jóvenes. Los blastos son de tamaño grande, aunque puede observarse algún elemento más pequeño, especialmente en SP. El núcleo es de perfil redondeado de cromatina laxa e inmadura, y el citoplasma, moderadamente abundante y basófilo, suele contener granulación azurófila o algún bastón de Auer. Ocasionalmente los blastos muestran granulación más grande (seudo-Chediak-Higashi) por fusión anómala de la granulación primaria.
Los precursores de los eosinófilos se hallan aumentados en médula ósea, pero estos no exhiben las alteraciones morfológicas y citoquímicas características de la LAM asociada a anomalías en el cromosoma 16. También suele detectarse un aumento de los basófilos.
Los blastos expresan antígenos mieloides y, de forma característica en esta entidad, coexpresan CD19. También son CD34 positivos. Los pacientes con este tipo de leucemia suelen alcanzar remisiones completas y una larga supervivencia con los esquemas terapéuticos adecuados.
Leucemia aguda mieloide con inv(16)/t(16;16)La LAM con inv(16)(p13q22) o t(16;16)(p13q11) y gen de fusión CBFβ/MYH11 constituye alrededor de un 5–8% de las LAM. Aunque puede presentarse a cualquier edad, predomina en pacientes jóvenes. Los blastos muestran diferenciación granulocítica y monocítica con morfología característica de la leucemia mielomonocítica (M4 según la clasificación FAB). Puede verse algún bastón de Auer ocasional. Se asocia a la presencia de eosinófilos anómalos en médula ósea. Morfológicamente, las anomalías en los eosinófilos se deben a la presencia de gránulos anormalmente grandes, que son de color violeta púrpura, e incluso pueden llegar a oscurecer de forma muy marcada a la célula. La reacción frente a la naftol-ASD-cloroacetatoesterasa, normalmente negativa en los eosinófilos, es positiva en los eosinófilos anormales. La reacción a las mieloperoxidasas (MPO) suele ser positiva. Los blastos monocíticos y los promonocitos muestran positividad para las estersasas, aunque puede ser más débil de lo esperado, o incluso la reacción puede ser negativa.
La mayoría de los casos muestran un inmunofenotipo complejo con poblaciones blásticas muy inmaduras CD34 y CD117 positivas, junto a blastos con diferenciación granulocítica que expresan CD13, CD33, CD65 y MPO o diferenciación monocítica que expresan CD14, CD4, CD11b, CD11c, CD64, CD36 y lisozima. Los pacientes de mayor edad tienen una supervivencia más corta. Por el contrario, los pacientes que presentan la alteración cromosómica adicional de una trisomía 22 tienen mejor pronóstico.
Leucemia aguda mieloide con t(15;17)En la LAM con t(15;17) (q22;q12) y gen de fusión PML-RAR-α existe un predominio de promielocitos atípicos que, morfológicamente, corresponden a la variedad M3 descrita por el grupo FAB.
Leucemia aguda mieloide con t(9;11)(p22;q23)La LAM con t(9;11)(p22;q23) y gen de fusión MLLT3-MLL se asocia a la presencia de blastos monocíticos (M4 o M5 según la clasificación FAB). Es más frecuente en niños (9–12% de las LAM en edad pediátrica y 2% de las LAM en adultos). Suele acompañarse de infiltración gingival o cutánea (sarcomas).
Los blastos son de tamaño grande, de moderada relación N/C, núcleo de perfil redondo de cromatina laxa e inmadura, con 1 o varios nucleolos prominentes, y citoplasma intensamente basófilo que puede contener una muy fina granulación azurófila y con presencia de mamelones o seudópodos. Con frecuencia se observan también promonocitos, cuyo citoplasma es menos basófilo y de granulación más abundante. Suelen mostrar positividad intensa a las esterasas y expresan marcadores monocíticos, tales como CD14, CD4, CD11b, CD11c, CD64, CD36 y lisozima. El pronóstico es intermedio.
Leucemia aguda mieloide con t(3;3) o inv(3)Es poco frecuente, ya que constituye el 1–2% de las LAM. Puede tratarse de una LAM de novo, o derivar de un síndrome mielodisplásico previo (SMD). Los blastos muestran una morfología característica de cualquiera de los subtipos FAB, excepto la M3, y más frecuentemente de M2, M4 o M7. Los pacientes suelen presentar anemia, y las plaquetas presentan cifras normales o elevadas. La observación de la SP, junto a la presencia de blastos, suele mostrar neutrófilos desgranulados y con seudo-Pelger, plaquetas gigantes e hipogranuladas y algún núcleo de megacariocito circulante.
En médula ósea existe un aumento de los megacariocitos de núcleos mono o bilobulados. Puede observarse displasia multilínea. Se asocia a monosomía 7 o del (5q). Muestra características agresivas y corta supervivencia.
Leucemia aguda mieloide con t(6;9)(p23;q34)La LAM con t(6;9)(p23;q34) y gen de fusión DEK-NUP214 es poco frecuente (<1–2%). A menudo se acompaña de basofilia (44–62% de los casos) y suele presentar las características morfológicas y citoquímicas de las variedades FAB M2 o M4. A menudo se asocia a pancitopenia y displasia, especialmente en la serie eritroide y granulocítica. En un tercio de los casos es posible la observación de bastones de Auer en los elementos blásticos. El inmunofenotipo de los blastos es mieloide con positividad para MPO, CD13, CD33, CD38 y HLA-DR. También pueden ser positivos para CD34, CD117 y CD15. Tienen un mal pronóstico.
Leucemia aguda mieloide con t(1;22)(p13;q13)Es poco frecuente (< 1% de las LAM). En general se asocia a una LA megacarioblástica en niños (sin Síndrome de Down), que en la mayoría de los casos tienen una edad inferior a un año. Se asocia a marcada hepatomegalia y esplenomegalia. Los pacientes muestran anemia, trombocitopenia y moderado aumento del recuento de leucocitos en sangre periférica. Los megacarioblastos son de tamaño moderado (12–18μm), elevada relación N/C, perfil nuclear redondeado o ligeramente irregular de cromatina laxa, reticulada con 1–3 nucleolos. El citoplasma es basófilo, a menudo agranular con característicos mamelones o seudópodos. Los blastos expresan CD41 y CD61. Los marcadores mieloides CD13 y CD33 pueden ser positivos, y son CD34 negativos. Responden a la quimioterapia intensiva.
Leucemias agudas mieloides con mutaciones genéticas:- •
Con mutación del gen FTL3.
- •
Con mutación del gen de la nucleofosmina (NPM1).
- •
Con mutación del gen CEBPA.
Ocurre en un 20–40% de las LAM. Se asocian a un mal pronóstico.
Leucemia aguda mieloide con mutación del gen de la npm1Constituye una tercera parte de las LAM. Se han descrito 40 mutaciones diferentes a nivel del exón 12 del gen. La mitad de ellas presentan un cariotipo normal. El pronóstico es bueno siempre y cuando no exista una mutación FTL3 asociada. Un 40% se asocian a dicha mutación, y tienen peor pronóstico. Es más frecuente en edades avanzadas y predomina en mujeres. Los pacientes pueden mostrar infiltración extramedular en encías, ganglios o piel. Suelen mostrar anemia, y cifras de leucocitos y plaquetas más elevadas que en la mayoría de LAM.
Existe una intensa asociación entre la mutación NPM1 y los subtipos morfológicos mielomonocíticos o monocíticos de LAM (M4, M5). El 80–90% de las LAM monocíticas presentan la mutación NPM1. La mutación también se ha descrito asociada a M1, M2 y M6.
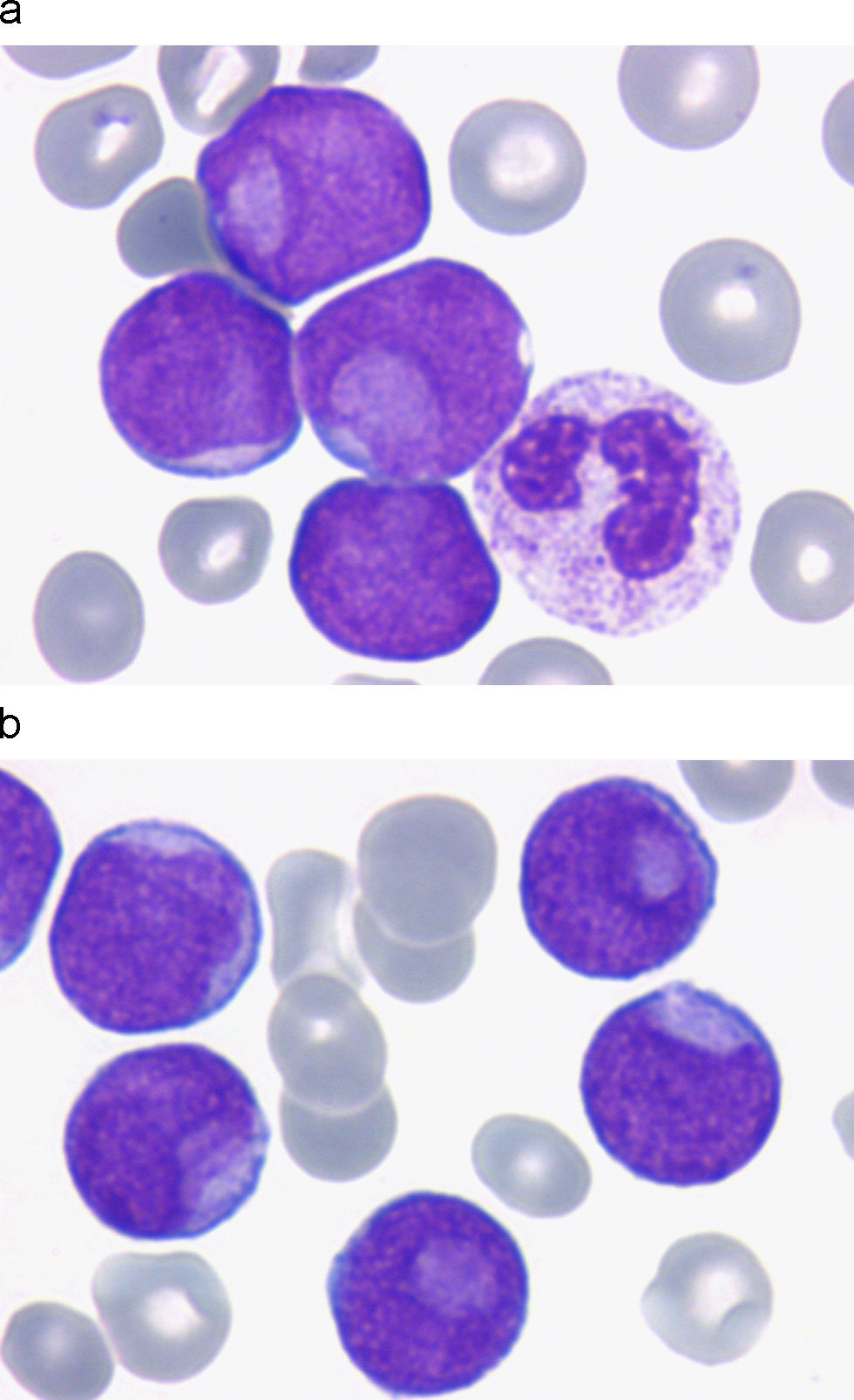
La morfología de los blastos es característica: aspecto mieloide, ocasionalmente de perfil irregular y de cromatina laxa e inmadura con depresión en forma de «huella digital», y citoplasma con esbozo de granulación (figs. 8a y b).
 Blastos mieloides en una LAM con mutación del gen NPM1. Obsérvese la característica depresión en el núcleo con forma de «huella digital».")
Desde el punto de vista inmunofenotípico además de los antígenos mieloides (CD33, CD13, MPO) los blastos de la LAM con mutación de la NPM1 expresan frecuentemente antígenos de diferenciación monocítica (CD14, CD11b). El diagnóstico se basa en la identificación de la alteración genética por técnicas de biología molecular, y por la detección inmunohistoquímica de la expresión de NPM en el citoplasma.
Leucemia aguda mieloide con mutación del gen cebpaFrecuencia: 6–15% de las LAM. Suele asociarse a valores elevados de hemoglobina, y a recuentos bajos de plaquetas. El cariotipo suele ser normal. Frecuente en el subtipo FAB M4. El pronóstico es favorable.
Leucemia aguda mieloide con displasiaEn la LAM con displasia se observa: a) Un 20% o más de blastos en sangre periférica o en médula ósea y b) Signos displásicos en más de un 50% de los elementos de al menos 2 líneas celulares.
Es frecuente el hallazgo de una pancitopenia severa. Puede presentarse como evolución de un síndrome mielodisplásico previo, o bien de novo. Los blastos generalmente son CD34 positivos, expresan antígenos mieloides (CD13 y CD33) y pueden expresar antígenos aberrantes (CD56 y/o CD7).
Leucemia aguda mieloide relacionada con tratamientos previosSe trata de una LAM que se presenta en pacientes que han recibido determinados tratamientos con agentes alquilantes, o bien con inhibidores de la topoisomerasa II. El efecto mutagénico de estos agentes provoca la aparición de la leucemia. Suelen verse signos displásicos en los neutrófilos (desgranulación y seudo-Pelger), así como diseritropoyesis. Algunos casos corresponden a una LAM sin maduración o, con menor frecuencia, a una LA mielomonocítica, monocítica, eritroleucemia o megacarioblástica. Los blastos suelen expresar el antígeno CD34 y antígenos mieloides (CD13, CD33). También pueden mostrar la expresión aberrante de CD56 y/o CD7. Es frecuente la presencia de anomalías cromosómicas complejas.
Leucemia aguda mieloide no categorizada previamenteEn este grupo se incluyen, entre otros, la mayoría de subtipos de LAM descritos por el grupo FAB y basados, fundamentalmente, en las características morfológicas de los elementos blásticos: LAM mínimamente diferenciada (LAM0), LAM sin maduración (LAM1), LAM con maduración (LAM2), LAM mielomonocítica (LAM4), LAM monoblástica (LAM5a) o monocítica (LAM5b), LA eritroide (LAM6), LA megacarioblástica (LAM7), LA basofílica, panmielosis aguda con mielofibrosis y sarcoma mieloide.
Leucemia aguda basofílicaLa incidencia de la LA basofílica es inferior al 1% de las LAM. Además de la infiltración medular, puede observarse afectación cutánea, hepato esplenomegalia, lesiones líticas óseas y síntomas relacionados con un aumento de las cifras sanguíneas de histamina. Los blastos son de mediano tamaño, elevada relación N/C y muestran un núcleo de perfil redondeado, oval o bilobulado de cromatina laxa y 1–3 nucleolos. El citoplasma es moderadamente basófilo y contiene un número variable de gránulos gruesos. Mediante microscopia electrónica de transmisión se observa que los gránulos gruesos contienen partículas electrón-densas características de los precursores de los basófilos o de las células cebadas. Este tipo de gránulos muestran positividad metacromática con azul de toluidina y también a las fosfatasas ácidas. Algunos blastos presentan positividad a «mazacotes» para el PAS. Son negativos para la MPO y para las esterasas inespecíficas. Los blastos expresan marcadores mieloides (CD13 y CD33), CD34 y HLA-DR.
Panmielosis aguda con mielofibrosisLa denominada panmielosis aguda con mielofibrosis es una proliferación mieloide que se acompaña de una intensa fibrosis de la médula ósea, con marcada pancitopenia y evolución rápidamente progresiva. Es una forma muy poco frecuente de LAM. Clínicamente suele presentarse con astenia intensa, fiebre y dolores óseos a los que se asocia una marcada pancitopenia, y la evolución es rápidamente progresiva. Cambios displásicos en las células mieloides son frecuentes y no se observan dacriocitos. Es muy difícil la obtención de material valorable mediante el aspirado medular, por lo que para el diagnóstico es imprescindible la biopsia ósea, junto a la realización de estudios inmunohistológicos. La biopsia es hipercelular y muestra una fibrosis difusa del estroma y un aumento de la proliferación de los precursores eritroides, granulocíticos y megacarioblásticos (panmielosis). Junto a ello, suelen observarse focos de células blásticas y megacariocitos displásicos. Los blastos expresan al menos uno o varios antígenos mieloides (CD13, CD33 y CD117), y son CD34 positivos.
El diagnóstico diferencial debe realizarse con otras LAM asociadas a fibrosis, como por ejemplo la leucemia aguda megacarioblástica, y con la mielofibrosis idiopática. Se asocia a muy mal pronóstico debido a su mala respuesta al tratamiento. La supervivencia suele ser de solo unos meses.
Sarcoma mieloideEl sarcoma mieloide consiste en una tumoración extramedular constituida por células mieloides inmaduras o mieloblastos. Es decir, se trata de una acumulación de blastos mieloides (con o sin maduración) en una localización diferente de la médula ósea. Aunque puede localizarse en cualquier lugar del cuerpo es más frecuente su aparición a nivel de piel, ganglios linfáticos, tracto gastrointestinal, hueso, tejido blando o testicular. Es más frecuente en varones y en las últimas décadas de la vida.
Morfológicamente el aspecto de las células del tumor corresponde a mieloblastos con o sin signos de maduración. Los blastos son, por tanto MPO positivos. En un 55% de los casos se demuestran alteraciones citogenéticas que incluyen monosomía 7, trisomía 8, inv(16) y otras. El diagnóstico diferencial debe realizarse con el linfoma maligno, y las pruebas histoquímicas e inmunofenotípicas demostrarán el origen mieloide de los blastos.
Leucemias agudas mieloides relacionadas con el síndrome de DownLos pacientes con síndrome de Down presentan un riesgo entre 10–100 veces superior a presentar una LAM. Antes de los 4 años el riesgo es 150 veces superior.
Un 10% de los recién nacidos con síndrome de Down muestran una mielopoyesis anormal indistinguible de una LAM megacarioblástica y que suele remitir espontáneamente, pero en un 13–30% de los casos progresa a una verdadera LAM en 1 a 3 años.
La LAM asociada al síndrome de Down en el 50% de los casos es del subtipo FAB M7, y generalmente se diagnostica antes de que el paciente cumpla los 3 años.
El estudio genético revela que en la LAM megacarioblástica del síndrome de Down, junto a la trisomía 21, se observa la mutación adquirida en GATA1. El pronóstico es más favorable que la LAM en niños sin síndrome de Down, debido a que presenta una mejor respuesta a la quimioterapia.
Neoplasia de células blásticas dendríticas plasmocitoidesProliferación maligna y agresiva de los precursores de las células dendríticas plasmocitoides, que se asocia con frecuencia a infiltración cutánea, de médula ósea y a diseminación leucémica. Su presentación es poco frecuente y aunque puede ocurrir a cualquier edad, la mayoría de los pacientes suelen tener entre 61–67 años. Las células proliferantes tienen un tamaño mediano, con un núcleo de perfil irregular y cromatina laxa e inmadura con 1 o varios nucleolos visibles. El citoplasma es basófilo y agranular.
Los blastos son negativos para la mieloperoxidasa y esterasas inespecíficas. Expresan CD4, CD43, CD56 y CD123. Positividad para cutaneous lymphocyte-associated antigen o TCL1, CLA.
Dos tercios de los pacientes muestran un cariotipo anormal. El curso clínico es agresivo con 12–14 meses de supervivencia.
