






Darunavir es uno de los fármacos inhibidores de la proteasa de última generación utilizado para el tratamiento de la inmunodeficiencia adquirida por VIH debido a su destacada eficacia terapéutica y su mejor tolerancia, entre otras peculiaridades. Varios estudios demuestran una correlación entre la dosis y el efecto de darunavir, aunque todavía no se ha establecido un intervalo terapéutico para las concentraciones del fármaco. Estos factores junto con una elevada variabilidad farmacocinética interindividual además de interacciones farmacológicas con otros fármacos, refuerzan la idea de la importancia de disponer de un método sensible para la monitorización de sus concentraciones plasmáticas en pacientes tratados.
ObjetivosValidación técnica de un método para la determinación de las concentraciones plasmáticas de darunavir mediante LC/MS/MS.
Materiales y métodosEl proceso de validación se desarrolló según el procedimiento descrito en la guía ICH Topic Q 2B Validation of Analytical Procedures: Methodology (CPMP/ICH/281/95). El darunavir se extrae del plasma mediante una precipitación de proteínas. La separación cromatográfica se consiguió utilizando una columna X-BridgeTM C18 3,5μm 2,1 × 100mm (Waters®) con un programa de elución en gradiente de acetonitrilo y tampón. Para la detección de los analitos, se utilizó un espectrómetro de masas de triple cuadrupolo Quattro micro con electroespray en modo de ionización positivo.
ResultadosLinealidad (0,1-10μg/mL): y=18,85×-150,4 r2: 0,997. Precisión: CV intraensayo: 1,07-4,62%. CV interensayo: 2,72-4,70%. Exactitud: intra-ensayo: 1-9%; inter-ensayo: 2-7%; LD: 0,05μg/mL; LQ: 0,15μg/mL.
ConclusionesEste método desarrollado basado en LC/MS/MS, posee una adecuada sensibilidad y reproducibilidad para la determinación de darunavir en plasma de pacientes tratados.
Darunavir is a latest generation of protease inhibitors (PI) drugs used as a treatment of HIV infection owing to its improved efficacy and its better tolerance among other characteristics. Several studies have demonstrated a correlation between the dose and the effect of darunavir, although a therapeutic range for the drug concentrations has not yet been established. These factors, besides the high inter-individual variability and the adverse effects resulting from drug interactions, reinforce the need for having a sensitive method for monitoring its plasma concentrations in treated patients.
ObjectiveTechnical validation of a new method for darunavir plasma concentration monitoring by LC/MS/MS.
Materials and methodsThe method validation procedure was based on the recommendations published in the guidelines ICH Topic Q 2B Validation of Analytical Procedures: Methodology (CPMP/ICH/281/95). Darunavir was extracted from plasma samples by a protein precipitation procedure. The chromatographic separation was achieved with a gradient program (acetonitrile/buffer) on an X-BridgeTM C18 3.5μm 2.1×100mm (Waters®). Analytes quantification is performed by electrospray ionization in positive mode, a Quattro Micro triple quadrupole mass spectrometer.
ResultsLinearity (0.1-10μg/mL): y=18.85×-150.4 r2: 0.997. Precision: Intra-assay CV: 1.07-4.62%. Inter-assay CV: 2.72-4.70%. Accuracy: Intra-assay: 1-9%. Inter-assay: 2-7%. LOD: 0.05μg/mL, LLOQ: 0.15μg/mL.
ConclusionThe developed method based on LC/MS/MS has an adequate sensibility and reproducibility for the determination of plasma concentrations of darunavir in treated patients.
El tratamiento de la enfermedad producida por el VIH se fundamenta en una terapia combinada de varios fármacos antiretrovirales incluyendo generalmente dos análogos nucleosídicos de la transcriptasa inversa (INRT), junto con un inhibidor de la proteasa (IP) o un inhibidor no nucleosídico de la transcriptasa inversa (INNRT). En los últimos años se han desarrollado nuevas opciones terapéuticas que aportan una mayor eficacia y tolerancia y sobretodo con especial interés en el desarrollo de tratamientos dirigidos a pacientes que han tenido experiencia previa con tratamientos antiretrovirales y que han presentado resistencia a estos fármacos1.
Darunavir (DRV) es uno de los nuevos inhibidores de proteasa de última generación con potente actividad frente a diferentes cepas virales del VIH resistentes a otros IP. Darunavir recibe el nombre comercial de Prezista® y es utilizado en combinación con otros fármacos antiretrovirales en pacientes con experiencia previa de tratamientos antiretrovirales y en pacientes “naïve”.
Actividad antiviralDarunavir presenta en su estructura molecular un grupo 3(R), 3a(S), 6a(R)-bistetrahidrofuraniluretanoisóstero (fig. 1). Este grupo favorece la formación de fuertes enlaces de tipo puentes de hidrógeno con el centro activo de la proteasa. Este tipo de unión representa una asociación rápida con la enzima y también una disociación lenta teniendo como consecuencia una mayor eficacia y duración de acción contra la proteasa viral. La Kd (indicadora de la afinidad o fuerza de unión del enzima al sustrato) del darunavir es de Kd 4,5×10−12 mol/L, 100 veces superior al amprenavir (Kd 3,9×10−10 mol/L), otro fármaco inhibidor de la proteasa, el cual presenta una estructura química similar al darunavir2,3.
Propiedades farmacocinéticas, 3a(S), 6a(R)-bistetrahidrofuraniluretanoisóstero).")
Darunavir administrado conjuntamente con ritonavir por vía oral presenta una absorción rápida alcanzando concentraciones plasmáticas (Cmax) a las 2,5-4 horas. La biodisponibilidad oral absoluta se incrementa del 37% a un 82% cuando se combina con ritonavir (14 veces) y disminuye a un 30% cuando se administra con alimento.
En plasma se encuentra unido un 95% a la α-ácidoglicoproteína (AAG). El volumen de distribución también se incrementa en presencia de ritonavir (Vd: 131L) frente a darunavir administrado en monoterapia (Vd: 88L)4.
Darunavir sufre un metabolismo oxidativo hepático mediante el citocromo P450, principalmente el CYP3A4, dando lugar a tres metabolitos oxidativos con actividad 10 veces inferior al darunavir. El interés de su uso combinado con 100mg de ritonavir recae en el efecto inhibitorio de éste último sobre el metabolismo hepático de darunavir favoreciendo el incremento de la exposición sistémica de darunavir. Como consecuencia su semivida de eliminación se incrementa a 15 horas. La eliminación del fármaco intacto es del 41,2% en heces y un 7,7% en orina4.
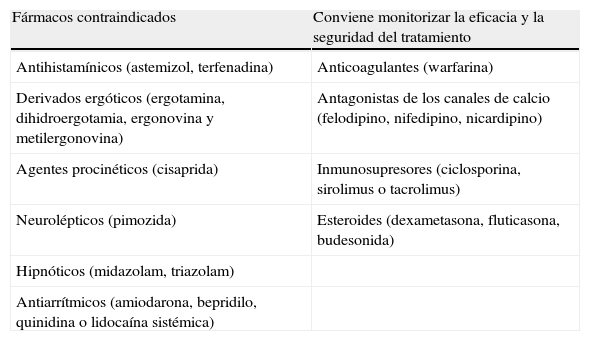
Interacciones farmacológicasDarunavir y ritonavir son inhibidores y sustratos del citocromo CYP3A4, por lo que la administración conjunta con otros fármacos que sean metabolizados por el mismo sistema dará lugar a alteraciones en las concentraciones plasmáticas de dichos fármacos afectando en la eficacia terapéutica o bien en la aparición de efectos adversos. Existen determinadas combinaciones con ciertos fármacos que están totalmente contraindicadas, mientras que hay otras asociaciones con darunavir que aunque no estén contraindicadas5, sí que requieren de una monitorización de las concentraciones plasmáticas de darunavir para tener un manejo controlado del régimen terapéutico que se ha instaurado (tabla 1).
Interacciones farmacológicas contraindicadas con darunavir
| Fármacos contraindicados | Conviene monitorizar la eficacia y la seguridad del tratamiento |
| Antihistamínicos (astemizol, terfenadina) | Anticoagulantes (warfarina) |
| Derivados ergóticos (ergotamina, dihidroergotamia, ergonovina y metilergonovina) | Antagonistas de los canales de calcio (felodipino, nifedipino, nicardipino) |
| Agentes procinéticos (cisaprida) | Inmunosupresores (ciclosporina, sirolimus o tacrolimus) |
| Neurolépticos (pimozida) | Esteroides (dexametasona, fluticasona, budesonida) |
| Hipnóticos (midazolam, triazolam) | |
| Antiarrítmicos (amiodarona, bepridilo, quinidina o lidocaína sistémica) |
En un ensayo clínico los estudios de la fase II se llevan a cabo para determinar la eficacia terapéutica de un fármaco en un grupo de pacientes afectados por dicha enfermedad. Esta fase se divide en la fase IIA en la que se estudia la dosificación y la fase II B en la que se llevan a cabo estudios para ver la eficacia de dicha dosificación ensayada. En esta fase se intentó buscar la dosis óptima de darunavir utilizando diferentes dosis y cambiando el régimen de la posología. Se demostró entonces que existía una relación proporcional entre la dosis y la exposición sistémica AUC24h (área bajo la curva), Cmin (concentración mínima), Cmax (concentración máxima), t1/2term (vida media del fármaco)6. Aunque no se ha establecido un intervalo terapéutico propiamente dicho, sí que se ha hallado un rango de concentraciones más eficaces y seguras para la Cmin referente a los pacientes participantes que tomaban una dosis de 600mg DRV/100mg ritonavir dos veces al día (Cmin: 1,3-7,4μg/mL) y con una dosis de 800mg/DRV/100mg ritonavir una vez al día (Cmin: 0,4-7,2μg/mL)4. La media de las Cmin fue 37 veces superior a la EC50=55 ng/mL4–6 lo cual significa que la Cmin no caía por debajo de los niveles de eficacia terapéutica. No obstante, no hay una buena correlación entre la respuesta virológica y la concentración de DRV considerada aisladamente, por lo que no se ha identificado un umbral de la concentración valle de DRV relacionado con una mayor probabilidad de respuesta virológica (concentración mínima efectiva [CME])7,8.
Sin embargo, sí que se encontró que el mejor predictor de la respuesta virológica era el cociente inhibitorio (IQ) ratio entre la Cmin y el valor de EC50 basal (concentración efectiva del 50% en pre-dosis), de manera que valores de IQ≥1,5 significaban una mayor probabilidad de reducción de la carga viral4.
Por otro lado, se demostró que existía una relación entre la dosis y el efecto de darunavir como también la eficacia superior de darunavir respecto a otros inhibidores de proteasa6,7.
Como conclusión en estos ensayos se identificó que la dosis óptima era la de 600mg de darunavir potenciado con 100mg de ritonavir dos veces/día6 y fue con esta dosis seleccionada con la que evualaron a posteriori la tolerancia y seguridad del fármaco. Se observó que darunavir era un fármaco bien tolerado, con reacciones adversas de grado 1 y 2, con una incidencia de reacciones adversas inferior al resto de IP4–9.
Otros estudios recientes también han demostrado que el tratamiento de darunavir en monoterapia presentaba una eficacia no inferior a la triple terapia usada hasta ahora formada por darunavir y dos análogos nucleósidicos de la transcriptasa reversa10. Estos resultados son relativamente novedosos, por lo que todavía quedan por realizar más estudios para ofrecer más información a tal evidencia; no obstante, estos hallazgos aportan matices de suma importancia en la terapia antiretroviral de la enfermedad producida por el VIH.
En conclusión, darunavir es un fármaco con una eficacia terapéutica destacada y una buena tolerancia. No obstante, dado que aún no se ha establecido un rango terapéutico bien definido de concentraciones plasmáticas; que su sistema de metabolización hepático es compartido por muchos otros fármacos (derivando así en interacciones farmacológicas); y que existe una elevada variabilidad farmacocinética interindividual, es importante disponer en el laboratorio clínico de un método sensible para la monitorización de sus concentraciones plasmáticas en aquellos pacientes tratados con posibles riesgos de desencadenar reacciones adversas o una disminución de la eficacia terapéutica del tratamiento. Por ello, el objetivo de este trabajo fue elaborar un método previo al estudio con pacientes tratados mediante LC/MS/MS para la determinación de darunavir en plasma.
Materiales y métodosLos ensayos para el desarrollo y validación del método para la determinación de darunavir tuvieron lugar en el Laboratorio de Farmacología de Fase I del Servicio de Bioquímica Clínica del Hospital Clínic de Barcelona durante un período de dos meses liderado por la Dra. Mercè Brunet.
Estándares y reactivosEl estándar de darunavir (DRV) fue suministrado por Tibotec-Johnson&Johnson y se almacenó a −20°C.
Ritonavir deuterado (A-86903), como estándar interno, fue suministrado por Abbot Diagnostics. Ácido fórmico (calidad para LC/MS) y acetato amónico (calidad para LC/MS) fueron obtenidos de Fluka (Steinheim, Alemania). Metanol (calidad para HPLC) y acetonitrilo (calidad para HPLC) fueron comprados a Panreac (Barcelona).
Soluciones estándar y curva de calibraciónSe prepararó una solución stock de darunavir de una concentración de 2mg/mL y del estándard interno (ritonavir deuterado) de 1mg/mL usando metanol como solvente. Las soluciones fueron almacenadas a −80°C para asegurar la estabilidad al menos durante 6 meses (según experiencia previa de los tests de estabilidad realizados en el laboratorio, cuyas áreas de dichas soluciones metanólicas congeladas eran comparables con disoluciones nuevas recién preparadas).
El pool de plasma de un grupo sano libre de fármaco fue proporcionado por el Banc de Sang i Teixits de Barcelona (BST Barcelona). Se analizó previamente como un blanco para comprobar la ausencia de picos de sustancias interferentes en el análisis cromatográfico, con el fin de utilizarlo como matriz biológica para calibradores y controles. Se prepararon 7 puntos de calibración (0,1μg/mL, 0,25μg/mL, 0,5μg/mL, 1μg/mL, 2,5μg/mL, 5μg/mL, 10μg/mL) y 4 concentraciones para los controles (0,3μg/mL, 0,9μg/mL, 3μg/mL, 7,5μg/mL) a partir de una solución de trabajo de 100μg/mL.
Preparación de la muestraEl primer paso consistió en añadir 50μl de calibrador o control en eppendorfs junto con 600μl de una solución de acetonitrilo/metanol (50:50) y 40μl de estántar interno (SI) de una solución de trabajo (10μg/mL en metanol/agua 50:50). Inmediatamente se agita al vórtex durante 30 segundos y se centrifugan los tubos en una centrífuga (Abbott) a 10.900rpm durante 10min a temperatura ambiente. A continuación 200μl del sobrenadante se transfieren a un vial de cromatografía donde previamente se ha añadido 200μl de agua de calidad Milli Q y se mantienen a 5°C en el interior del carro de muestras del cromatógrafo hasta su análisis11.
Instrumentación y condiciones analíticasEl cromatógrafo líquido de alta resolución (HPLC) utilizado fue el modelo Alliance 2795 (Waters, Milford, MA, USA). La columna analítica del HPLC fue una columna X-Bridge™ C18 (2,1 × 100mm, 3,5μm, Waters) que se mantenía a 65°C en el horno de la columna.
La fase móvil estaba compuesta de una solución A (acetonitrilo) y una solución B (acetato amónico 3mM/0,1M ácido fórmico) y seguía un programa de elución en gradiente. Se iniciaba con un 60% de fase móvil A y un 40% de fase móvil B durante 1,5min. A continuación durante 0,5min se incrementaba el flujo al 100% de la fase móvil A y se mantenía durante 1,5 minutos. Finalmente durante 0,1min descendía a un 60% de fase móvil A y a un 40% de fase móvil B manteniéndose en esta proporción durante 1,9min (fig. 2). El volumen de inyección era de 40μl, el flujo de 0,4mL/min y un tiempo total de análisis de 5,5min.
 para el análisis cromatográfico de darunavir.")
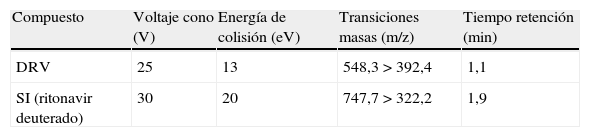
Para la detección, se utilizó un espectrómetro de masas de triple cuadrupolo Quattro micro (Micromass, Manchester, UK). La ionización se producía por electrospray en modo positivo usando un potencial del capilar de 1kV. La temperatura de la fuente era de 120°C y la temperatura de desolvatación de 300°C. El flujo del gas de nebulización era de 700 l/h y el flujo del gas de cono de 50 l/h. Las transiciones m/z de los iones detectados fueron 548,3>392,4 para el darunavir y 747,7>322,2 para el ritonavir deuterado (ya que son éstos los fragmentos hijos mayoritarios tras la fragmentación de la molécula tal y como se describe en otros métodos descritos en la bibliografía)11–13. Dichas transiciones se han escogido con una resolución de 0,6 (anchura del pico a mitad de altura). En la tabla 2 se recogen parámetros específicos para cada transición estudiada. El procesamiento de los datos se realizó usando el software MassLynx™ V 4.0 y la curva de calibración se construyó utilizando un factor de 1/x para ponderar la recta calculada por mínimos cuadrados.
Parámetros específicos del detector de espectrometría de masas y características del análisis cromatográfico de los analitos
| Compuesto | Voltaje cono (V) | Energía de colisión (eV) | Transiciones masas (m/z) | Tiempo retención (min) |
| DRV | 25 | 13 | 548,3 > 392,4 | 1,1 |
| SI (ritonavir deuterado) | 30 | 20 | 747,7 > 322,2 | 1,9 |
El proceso de validación se realizó siguiendo el protocolo descrito en la guía ICH Topic Q 2B Validation of Analytical Procedures: Methodology14.
En el caso de la precisión intra-día y exactitud, se realizaron 5 replicados para cada concentración de control durante el mismo día. En el caso de la exactitud y precisión inter-día, se realizaron 5 replicados diarios durante tres días diferentes. La precisión se expresó como desviación estándar relativa para cada control y no debía sobrepasar el 15% según indica la guía. La linealidad se evaluó en un rango de concentraciones entre 0,1-10μg/mL usando 7 concentraciones. Para aceptar la validación se asumió un valor mínimo de r2=0,997. Para aceptar el test de exactitud se consideraron valores óptimos el 100±15% del valor teórico del control según indica la guía. En el caso del límite de cuantificación (LQ) se analizaron 5 plasmas libres de fármaco a los cuales se les añadió una concentración de darunavir igual a la del calibrador más bajo (0,1μg/mL). La exactitud del LQ debería estar en un 100±20% y la precisión por debajo del 20%. En cuanto al límite de detección, se analizaron 5 plasmas libres de fármaco a los cuales se les había añadido una concentración de darunavir inferior a la concentración del calibrador más bajo (0,05μg/mL).
El efecto matriz se evaluó en las cuatro concentraciones de controles. Para ello, se añadió darunavir en concentraciones equivalentes a las obtenidas para los controles a cinco sobrenadantes resultantes de la extracción de plasmas blanco (sin fármaco) para cada concentración de control. Por otro lado, se prepararon soluciones estándar en fase móvil que contenían darunavir a concentraciones equivalentes. El resultado se obtuvo del ratio obtenido entre la media de las áreas de los cinco picos de darunavir de los blancos post-extracción y la media de las áreas de los estándares en fase móvil.
Existen diferentes maneras de evaluar la recuperación de un método, en nuestro laboratorio se ensayó para las cuatro concentraciones de controles comparando la media de las áreas de 5 replicados de cada concentración de control pre-extracción con la media de las áreas de los picos post-extracción.
El análisis del carryover se realizó según las recomendaciones de la guía Proposals for the description and measurement of carry-over effects in clinical chemistry15.
El análisis de los resultados de la validación se realizó con el programa de procesamiento de datos Excel.
ResultadosLos parámetros del espectrómetro de masas se optimizaron para favorecer las transiciones que presentaron mayor sensibilidad: 548,3>392,4 para el darunavir y 747,7>322,2 para el ritonavir deuterado. Las condiciones cromatográficas mostraban un perfil como el que se observa en la figura 3. Darunavir llega al detector en el minuto 1,1 y el ritonavir deuterado en el minuto 1,95 (tabla 2).
Validación
Se analizaron 6 muestras de plasma libres de darunavir y no se observaron interferencias con el análisis de darunavir. El método fue lineal en el rango de concentraciones de 0,1-10μg/mL obteniendo un valor de r2=0,997. Todos los puntos de la calibración presentaron una exactitud por encima del 85% (Intra-día: 92,5-97,3% e Inter-día: 92,5-97,74%) y una precisión por debajo del 15% (Intra-día: 1,9-3,3% e Inter-día: 0,7-1,2%) tal y como se indica en las guías para su análisis. En el caso del límite de cuantificación (LQ), se aceptó la concentración de 0,15μg/mL y para el límite de detección (LD) de 0,05μg/mL. En todos los casos el valor de recuperación estaba por encima del 75%. La evaluación del efecto matriz descartaba interferencias con darunavir y mostraba que las condiciones del análisis no afectaban a la ionización de la molécula. Todos los resultados de la validación se recogen en la tabla 3. El método se utilizó para monitorizar pacientes tratados con dicho fármaco. Se evaluaron las posibles interacciones con otros fármacos antiretrovirales (raltegravir, ritonavir, lopinavir, atazanavir), inmunosupresores (tacrolimus, ciclosporina, everolimus y sirolimus) y sus metabolitos y no se detectaron interferencias analíticas.
Valores de precisión intra-día e inter-día (R.S.D %) y exactitud de darunavir en plasma (n=5)
| Controles | Concentración (μg/mL) | Precisión (%) | Exactitud (%) | ||
| Intra-ensayo | Inter-ensayo | Intra-ensayo | Inter-ensayo | ||
| C1 | 0,3 | 3,3 | 1,2 | 92,6 | 92,62 |
| C2 | 0,9 | 2,7 | 1,04 | 92,5 | 92,51 |
| C3 | 3 | 2,7 | 1,17 | 97,3 | 97,6 |
| C4 | 7,5 | 1,9 | 0,70 | 95,2 | 97,74 |
| LD | 0,05 | ||||
| LQ | 0,15 | ||||
El uso de los inhibidores de proteasa en el régimen terapéutico del tratamiento de la inmunodeficiencia adquirida por VIH se está convirtiendo cada vez más en una práctica eficaz a la hora de tratar a estos pacientes afectados por el virus.
Sobretodo, los IP adquieren mayor importancia en aquellos pacientes en los que una terapia previa con otros tratamientos antiretrovirales ha fracasado debido a cepas del virus resistente a estos fármacos, y entre ellos se encuentra darunavir, uno de los nuevos fármacos con excelente eficacia antiretroviral y una buena tolerancia demostrada. No obstante, todavía se desconocen algunos aspectos en cuanto a las concentraciones asociadas a un rango terapéutico establecido de darunavir o el comportamiento farmacocinético en determinadas poblaciones. Por ello, la monitorización de sus concentraciones plasmáticas resulta de interés para el manejo de efectos adversos, para optimizar la eficacia del tratamiento o para controlar determinadas interacciones farmacológicas.
Estos intereses nos condujeron a desarrollar un método de determinación de darunavir en muestras plasmáticas mediante cromatografía líquida acoplada a espectrometría de masas. El método constó de una primera parte de tratamiento previo de la muestra plasmática mediante precipitación de proteínas, una metodología fácil de llevar a cabo y poco costosa que mostró tener una elevada fiabilidad y reproducibilidad. La adición de un tampón formado por acetato amónico 3mM/ácido fórmico 0,1M (60:40) a la fase móvil favoreció la producción de formas iónicas de los analitos (M+H+) y en menor proporción (M+NH4+). Los fragmentos hijos obtenidos fueron detectados en el espectrómetro de masas mediante ionización en electrospray en modo positivo (ESI+) ya que se obtenían mejores resultados que mediante electrospray en modo negativo (ESI-).
Respecto a la validación, el método fue lineal en todo el rango de concentración de la curva de calibración (0,1-10μg/mL). Este rango de concentraciones se obtuvo de los valores más altos encontrados de darunavir en estudios clínicos y farmacocinéticos.
La precisión y exactitud intra-día e inter-día resultaron satisfactorios. La ausencia de picos en el cromatograma mostró que no había interferencias con analitos de la propia matriz biológica ni con otros fármacos y por lo tanto reflejaba que el método tenía una buena especificidad. Por otro lado los ensayos de precisión y exactitud también demostraron que las condiciones de análisis son las adecuadas para una correcta ionización de la molécula. El método además presentaba una buena sensibilidad basándose en un LD de 0,05μg/mL y un LQ de 0,15μg/mL.
En conclusión, el método ensayado reúne todos los requisitos respecto a sensibilidad, especificidad, precisión y exactitud, que son necesarios para poder realizar una correcta determinación de las concentraciones plasmáticas de darunavir y poder aplicarlo a su monitorización farmacológica en aquellas poblaciones especiales que lo requieran.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nos gustaría agredecer a Tibotec-Johnson&Johnson el suministro de darunavir sustancia pura.
Este trabajo corresponde a una comunicación científica presentada y premiada en el III Congreso Nacional del Laboratorio Clínico celebrado en Valencia del 14 al 16 de octubre de 2009.
