



La ceftazidima, el meropenem y la piperacilina son antibióticos β-lactámicos de amplio espectro empleados en el tratamiento empírico de pacientes críticos con sepsis. Estos fármacos presentan una actividad antimicrobiana dependiente del tiempo por lo que sus concentraciones de masa en el plasma deberían medirse y mantenerse por encima de la concentración mínima inhibitoria. El objetivo de este estudio es desarrollar y validar un procedimiento de medida basado en la cromatografía de alta y rápida resolución acoplada a la espectrometría de masas en tándem para la medición simultánea de la concentración de masa de ceftazidima, meropenem y piperacilina en el plasma.
Material y métodosDespués de una precipitación de proteínas de las muestras con acetonitrilo y posterior dilución con agua, los eluatos son introducidos en una columna C18 de fase inversa usando un gradiente de agua/acetonitrilo que contiene ácido fórmico. Los antibióticos son detectados mediante un espectrómetro de masas de triple cuadrupolo trabajando en las modalidades de ionización por electroespray y monitorización de reacción múltiple.
ResultadosLos límites de cuantificación son cercanos a 0,50mg/l. Los coeficientes de variación y sesgos relativos son inferiores a 10,8 y 12,0%, respectivamente. Los valores de recuperación están comprendidos entre 55,7 y 77,4%. La evaluación del efecto matriz muestra una sobreexpresión iónica para todos los antibióticos. No se observan interferencias ni contaminación por arrastre.
ConclusionesEl procedimiento de medida validado podría ser empleado en la práctica diaria del laboratorio clínico para la medición de estas magnitudes farmacológicas, principalmente en pacientes críticos con sepsis.
Ceftazidime, meropenem and piperacillin are broad spectrum antibiotics often used for the empirical treatment of infections in critically ill patients with sepsis. These antibiotics show time-dependent antimicrobial activity, meaning that the antibiotic mass concentration in plasma should be measured and maintained above the minimal inhibitory concentration. The aim of this study was to develop and to validate an ultra-performance liquid chromatography-tandem mass spectrometry procedure for the simultaneous measurement of ceftazidime, meropenem, and piperacillin mass concentration in plasma.
Material and methodsAfter protein precipitation with acetonitrile and subsequent dilution of the supernatant with water, eluates were introduced into a reverse-phase C18 column using a water/methanol gradient containing formic acid. Antibiotics were detected by electrospray ionisation mass spectrometry in multiple reaction monitoring mode.
ResultsThe lower limits of quantification were close to 0.50mg/l. Coefficients of variation and absolute relative biases were less than 10.8 and 12.0%, respectively. Recovery values ranged from 55.7 to 77.4%. Evaluation of the matrix effect showed ion enhancement for all antibiotics. No interferences or carry-over were observed.
ConclusionsThe validated measurement procedure could be used in daily clinical laboratory practice to measure the mass concentration of these antibiotics in plasma, and in critically ill patients with sepsis.
La ceftazidima (fig. 1A), el meropenem (fig. 1B) y la piperacilina (fig. 1C) son antibióticos β-lactámicos ampliamente utilizados en el tratamiento empírico de pacientes críticos con sepsis1–3. En este tipo de pacientes, la farmacocinética de estos antibióticos es heterogénea e impredecible dadas las características físico-patológicas diferenciales que presentan respecto a otras poblaciones (elevada variabilidad en los volúmenes de distribución, eliminación, penetración y distribución del fármaco en los tejidos, así como la presencia de hipoalbuminemia y disfunción orgánica). Por este motivo, se recomienda que los regímenes posológicos en estos pacientes se basen en conceptos farmacocinéticos (PK) y farmacodinámicos (PD). Para la ceftazidima (CFT), el meropenem (MEM) y la piperacilina (PIP), al tratarse de antibióticos dependientes del tiempo (con mínimo o nulo efecto postantibiótico), el parámetro PK/PD que mejor se asocia con la eficacia del tratamiento y con la prevención de la aparición de resistencia bacteriana es el porcentaje de tiempo durante el cual la concentración del antibiótico en el plasma se mantiene por encima de la concentración mínima inhibitoria (CMI) (% T>CMI o % fT>CMI)4–7. Sin embargo, actualmente no está claro cuáles han ser los valores de estos parámetros PK/PD. Mientras que algunos estudios sugieren que estos valores deben estar comprendidos entre el 40 y el 70%, otros recomiendan aumentarlos hasta el 100% o, incluso, que los valores de la concentración del antibiótico en el plasma se mantengan entre cuatro y cinco veces por encima de la CMI4–7. La posibilidad de ajustar la dosis de estos antibióticos, a partir de los valores obtenidos mediante la medición de sus concentraciones de masa en el plasma, podría suponer un avance importante en el tratamiento de las infecciones graves en los pacientes críticos.
 ceftazidima, B) meropenem y C) piperacilina.")
En la actualidad, existen pocos trabajos publicados en los que se haya estudiado la medición simultánea de estas magnitudes farmacológicas mediante la cromatografía líquida de alta y rápida resolución (UHPLC) acoplada a la espectrometría de masas en tándem (MS/MS)8–12. Tres de ellos9–11 presentan tiempos cromatográficos relativamente elevados, y otros tres emplean tratamientos o preparación de la muestra que requieren tiempos considerables10–12.
El objetivo de este trabajo es desarrollar y validar un procedimiento de medida para la medición simultánea de la concentración de masa de ceftazidima, meropenem y piperacilina en el plasma basado en la UHPLC-MS/MS, que requiera un proceso de preparación de la muestra sencillo y práctico, así como un tiempo cromatográfico reducido.
Material y métodosReactivos químicosPara la preparación de los materiales de calibración y de control se emplean los materiales de referencia certificados Ceftazidime CRS (pureza del 85,3%), Meropenem trihydrate CRS (pureza del 87,0%) y Piperacillin CRS (pureza del 94,4%) de la Farmacopea Europea (European Directorate for the Quality of Medicines-Council of Europe, Estrasburgo, Francia). Como patrones internos (PI) se utilizan los fármacos deuterados D5-Ceftazidima (PI para la CFT), D6-Meropenem (PI para el MEM) y D5-Piperacilina (PI para la PIP), que se obtienen de la empresa Toronto Research Chemicals Inc (Ontario, Canadá). El acetonitrilo, el agua, el dimetilsulfóxido (DMSO) y el ácido fórmico, todos ellos de calidad LC-MS, son proporcionados por Sigma-Aldrich S. A. (Madrid, España).
Preparación de soluciones primarias y soluciones de trabajoSe preparan dos soluciones primarias de 2,0g/l que contienen los fármacos en estudio, una para los materiales de calibración y otra para los materiales de control. Las soluciones primarias se obtienen pesando una cantidad apropiada de cada material de referencia certificado, y disolviendo todos estos materiales en 20mL de una solución compuesta por agua: metanol: DMSO (50:25:25, v/v/v). Seguidamente, se elaboran diferentes soluciones acuosas secundarias (9 para los materiales de calibración y 3 para los materiales de control) que presentan 10 veces el valor teórico de los materiales de calibración y de control que se emplearán en el estudio. Estas soluciones se almacenan a -80°C en alícuotas de 50μL en tubos tipo Eppendorf. En el momento del análisis, se preparan los materiales de calibración (0,0; 0,50; 1,00; 5,00; 15,0; 45,0; 75,0; 125 y 175mg/l) y los materiales de control (3,00; 30,0 y 120mg/l) añadiendo una parte de volumen de la correspondiente solución secundaria descrita anteriormente sobre 9 partes de volumen de un material de blanco (una mezcla de 30 muestras de plasmas de pacientes que no han ingerido ni se les ha suministrado ninguno de los fármacos en estudio).
Por otro lado, se preparan tres soluciones primarias, una para cada PI, diluyendo 1mg del PI en 10mL de un disolvente apropiado (DMSO, metanol o agua, de acuerdo con las instrucciones del certificado de análisis del fabricante). Estas soluciones se almacenan a -80°C en alícuotas de 100μL en tubos tipo Eppendorf. En el momento del análisis, y para 20 muestras, se prepara una solución de trabajo de PI añadiendo 100μL de cada uno de los PI en 6mL de acetonitrilo.
Preparación y tratamiento de las muestrasLa preparación y tratamiento de las muestras consiste en una precipitación de proteínas seguida de una dilución acuosa del sobrenadante obtenido.
En un tubo tipo Eppendorf de 1,5mL, se añaden 100μL de cada calibrador, control o muestra de plasma de paciente y 300μL de la solución de trabajo de PI. La mezcla se agita en un vórtex durante 3min y, posteriormente, el tubo se centrifuga durante 10min a 11.000g a 4°C. Finalmente, 100μL del sobrenadante se diluyen con 400μL de agua, se agita la solución durante 5 s y se añade el contenido en un vial cromatográfico específico para su procesamiento.
Sistema de medidaSe emplea un sistema de medida ACQUITY®-UPLC® acoplado a un espectrómetro de masas ACQUITY®-TQD®, ambos de Waters Cromatografía S. A. (Sant Cugat del Vallès, España).
Condiciones cromatográficasLa separación cromatográfica de los componentes de la muestra se realiza a 30°C utilizando una columna analítica C18 en fase reversa Acquity UPLC® BEH™ 2,1×100mm; 1,7μm, equipada con un filtro de 0,2μm y una precolumna Acquity® UPLC® BEH™ C18 VanGuard Pre-column (5mm×2,1mm; 130Å, 1,7μm) (Waters Cromatografía S. A.). La fase móvil está compuesta por una solución de ácido fórmico 0,1% (v/v) en agua (fase móvil A) y una solución de ácido fórmico 0,1% (v/v) en acetonitrilo (fase móvil B). El flujo aplicado se mantiene a 0,4mL/min durante todo el proceso cromatográfico. Del minuto 0,0 al 0,5 se realiza una elución isocrática con una proporción de la fase móvil A del 98%. Seguidamente, entre los min 0,5 y 2,0, la proporción de la fase móvil A decrece gradualmente del 98 al 50% (gradiente lineal). Finalmente, se reequilibra la columna analítica durante 1,0min empleando la proporción inicial de la fase móvil A (98%). Las muestras extraídas se mantienen a (4±1) °C en el interior del muestreador y el volumen de inyección de muestra es de 10μL.
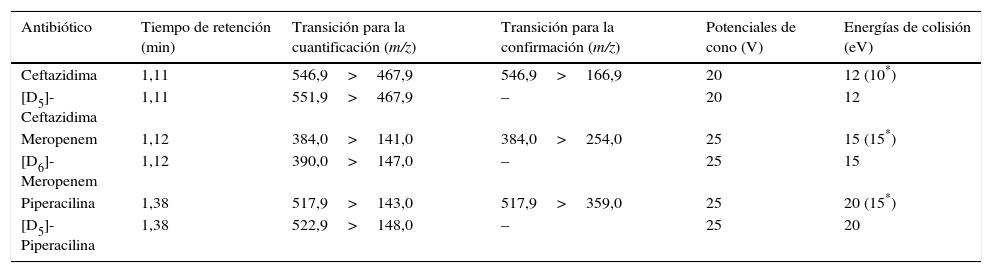
Condiciones del espectrómetro de masasEl espectrómetro de masas opera en las modalidades de ionización positiva mediante electroespray (ESI+) y de reacción de monitorización múltiple (MRM). Se utiliza nitrógeno como gas de nebulización y de desolvatación, y argón como gas de colisión. Para cada antibiótico, se emplean dos transiciones MRM: una para la cuantificación («quantifier») y otra para la identificación o confirmación («qualifier»). En cambio, para cada PI, se utiliza una única transición MRM. Las transiciones de MRM utilizadas, así como los potenciales de cono y las energías de colisión se muestran en la tabla 1. Además, para todos los antibióticos y sus correspondientes PI, se emplean los siguientes valores de los parámetros del espectrómetro de masas: un potencial de capilaridad de 1,2kV, un potencial de extracción de 3V, un potencial de radiofrecuencia de 0,1V, una temperatura de la fuente de ionización de 130°C, una temperatura de desolvatación de 450°C, un flujo de desolvatación de 800 L/h, un flujo del gas de colisión de 0,20mL/min y un tiempo de integración («Dwell time») de 50ms.
Parámetros del espectrómetro de masas para la ceftazidima, el meropenem, la piperacilina y sus correspondientes patrones internos
| Antibiótico | Tiempo de retención (min) | Transición para la cuantificación (m/z) | Transición para la confirmación (m/z) | Potenciales de cono (V) | Energías de colisión (eV) |
|---|---|---|---|---|---|
| Ceftazidima | 1,11 | 546,9>467,9 | 546,9>166,9 | 20 | 12 (10*) |
| [D5]-Ceftazidima | 1,11 | 551,9>467,9 | – | 20 | 12 |
| Meropenem | 1,12 | 384,0>141,0 | 384,0>254,0 | 25 | 15 (15*) |
| [D6]-Meropenem | 1,12 | 390,0>147,0 | – | 25 | 15 |
| Piperacilina | 1,38 | 517,9>143,0 | 517,9>359,0 | 25 | 20 (15*) |
| [D5]-Piperacilina | 1,38 | 522,9>148,0 | – | 25 | 20 |
ESI: electrospray ionization (ionización mediante electroespray); MRM: Multiple Reaction Monitoring (reacción de monitorización múltiple); m/z: relación masa-carga.
El procedimiento de desarrollo y validación está basado, principalmente, en la guía de la Agencia Europea del Medicamento (EMA)13 y en las recomendaciones del CLSI14,15.
Procedimiento de calibraciónCada vez que se realizan mediciones de las magnitudes farmacológicas se procesan los nueve materiales de calibración y los tres materiales de control.
La integración de las áreas bajo la curva de los picos cromatográficos suavizados y el cálculo de los valores de la concentración de masa de los antibióticos en el plasma se llevan a cabo mediante el programa informático TargetLynx® v. 4.1 de Waters Cromatografía. Las curvas de calibración se obtienen mediante un ajuste lineal por ponderación 1/X, excluyendo la opción de forzar la intersección de la recta por la ordenada en el origen.
SelectividadPara el estudio de la selectividad se procesan 10 muestras de pacientes que no están en tratamiento con los antibióticos en estudio pero sí con otros fármacos: antiepilépticos (fenitoína, lamotrigina, valproato), digoxina, micofenolato, u otros antibióticos (aztreonam, cefepina, cloxacilina, tobramicina, vancomicina). Todos los pacientes presentan unos valores de la concentración de masa del fármaco en el plasma dentro del intervalo terapéutico, o bien por encima de la concentración mínima inhibitoria del patógeno en cuestión.
Contaminación por arrastrePara el estudio de la contaminación por arrastre se procesan, en una misma serie y en dos días distintos, una muestra con un valor elevado (calibrador 8; 175mg/l) seguida de tres materiales de blanco (calibrador 0; 0mg/l).
Límite de cuantificaciónLa EMA13 define el límite de cuantificación (LLOQ) como el mínimo valor de una magnitud que puede obtenerse con una imprecisión aceptable (CV ≤ 20%) y cuya relación señal/ruido (S/N) es≥5.
Para estimar el LLOQ, se diluye un material de valor bajo (calibrador 2; 1,00mg/l) en una proporción 1:1 y en una proporción 1:4 con un material de blanco (calibrador 0; 0mg/l). Cada una de las diferentes soluciones diluidas, así como el material sin diluir, se procesan 10 veces en un único día y 10 veces durante 20 días no consecutivos utilizando una única serie por día.
Imprecisión y sesgoPara estimar la imprecisión y el sesgo se utilizan los tres materiales de control preparados anteriormente (3,0, 30 y 120mg/l). Para el cálculo de la imprecisión y sesgo intraseriales se procesan 10 veces, en una única serie y en un mismo día, los tres materiales de control. Por otra parte, para el cálculo de la imprecisión y sesgo interdiarios, se procesan las mismas muestras 10 veces, durante 20 días no consecutivos, utilizando una única serie por día. Los valores convencionales empleados para estimar los sesgos corresponden a los valores asignados mediante pesada.
Eficiencia del proceso cromatográfico (recuperación y efecto matriz)El estudio de la eficiencia del proceso cromatográfico está basado en la guía de la EMA13, en las recomendaciones del CLSI14,15 y en el procedimiento descrito por Viswanathan et al.16. Para llevarlo a cabo, se utilizan tres tipos de muestras diferentes a un valor de 3,00; 30,0 y 120mg/l para cada antibiótico y a un valor de 2,0mg/l, para los PI. Para cada uno de los antibióticos y PI, estos tres tipos de muestras son: soluciones primarias de CFT, MEM, PIP y de sus correspondientes PI diluidas con la fase móvil A (muestras A); seis muestras de plasma de pacientes diferentes que no contienen ninguno de los antibióticos y a las que se les añade CFT, MEM, PIP o PI después del proceso de extracción (muestras B); y las mismas 6 muestras a las que se les adiciona CFT, MEM, PIP o PI antes del proceso de extracción (muestrasC).
A partir de las áreas de los picos cromatográficos obtenidos después de procesar aleatoriamente todas estas muestras, el porcentaje de recuperación (RE) y del factor de efecto matriz (ME) para cada antibiótico se calculan como:
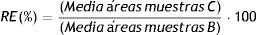

Así mismo, para conocer si los PI son capaces de compensar el posible efecto matriz ocasionado en la fuente de ionización, se calcula el porcentaje del factor de efecto matriz normalizado (n-ME) a partir de la siguiente fórmula:
Estabilidad
Se lleva a cabo un estudio de la estabilidad a corto (a los 1, 3 y 7 días a 5±3°C) y a largo plazo (a los 6 meses a -80°C) empleando los materiales de control, y un estudio de la estabilidad en el muestreador (a las 6, 12 y 24 h a 4±1°C) utilizando los extractos de los materiales de control.
Para cada estudio y magnitud farmacológica, se procesan las distintas muestras 10 veces y se evalúa la estabilidad calculando la diferencia relativa porcentual (PD) utilizando la siguiente ecuación:
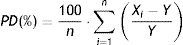
Donde Xi es cada uno de los valores obtenidos para las diferentes muestras utilizadas una vez trancurrido el período de almacenamiento de las mismas, Y es el valor teórico asignado, y n es el número de veces que se han procesado las muestras (n=10, en nuestro caso).
Según la EMA, se considera que las magnitudes son estables si la PD obtenida está comprendida entre±15%.
Aplicación en muestras biológicasEl procedimiento basado en la UHPLC-MS/MS descrito está siendo empleado en un programa de optimización de la terapia antibiótica en pacientes críticos, que ha sido aprobado por el Comité de Ética de nuestro hospital y cumple con los requisitos de la Asociación Médica Mundial y la Declaración de Helsinki.
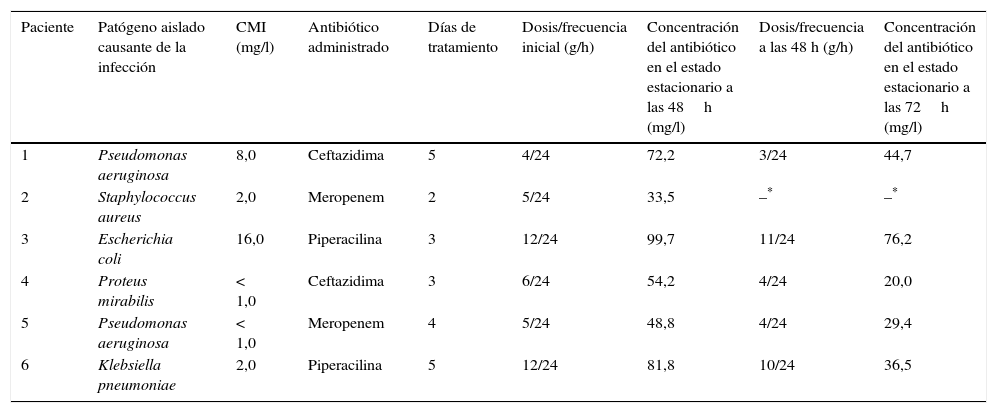
PacientesSe evalúa la aplicabilidad del procedimiento UHPLC-MS/MS procesando diferentes muestras de pacientes ingresados en el Servicio de Medicina Intensiva de nuestro hospital. Estos pacientes sufren algún tipo de infección bacteriana grave y son tratados con alguno de los antibióticos incluidos en el presente estudio (tabla 2).
Información relacionada con pacientes ingresados en el Servicio de Medicina Intensiva de nuestro hospital que presentan algún tipo de infección bacteriana
| Paciente | Patógeno aislado causante de la infección | CMI (mg/l) | Antibiótico administrado | Días de tratamiento | Dosis/frecuencia inicial (g/h) | Concentración del antibiótico en el estado estacionario a las 48h (mg/l) | Dosis/frecuencia a las 48 h (g/h) | Concentración del antibiótico en el estado estacionario a las 72h (mg/l) |
|---|---|---|---|---|---|---|---|---|
| 1 | Pseudomonas aeruginosa | 8,0 | Ceftazidima | 5 | 4/24 | 72,2 | 3/24 | 44,7 |
| 2 | Staphylococcus aureus | 2,0 | Meropenem | 2 | 5/24 | 33,5 | –* | –* |
| 3 | Escherichia coli | 16,0 | Piperacilina | 3 | 12/24 | 99,7 | 11/24 | 76,2 |
| 4 | Proteus mirabilis | < 1,0 | Ceftazidima | 3 | 6/24 | 54,2 | 4/24 | 20,0 |
| 5 | Pseudomonas aeruginosa | < 1,0 | Meropenem | 4 | 5/24 | 48,8 | 4/24 | 29,4 |
| 6 | Klebsiella pneumoniae | 2,0 | Piperacilina | 5 | 12/24 | 81,8 | 10/24 | 36,5 |
CMI: concentración mínima inhibitoria.
De acuerdo con los criterios del clínico, la estrategia terapéutica consiste en la infusión continua de alguno de los antibióticos en estudio, previa administración de una dosis de carga (infusión durante 30min) seguida de una dosis de mantenimiento (tabla 2). Asimismo, si a las 48h del inicio del tratamiento los valores de las diferentes magnitudes farmacológicas eran superiores a 4 veces la CMI, la dosis del fármaco fue reducida según el criterio del clínico (tabla 2).
Recogida de la muestraLas muestras de los pacientes se obtienen pasadas 24–48h del inicio del tratamiento antibiótico para garantizar que la concentración del fármaco se encuentra en las condiciones de estado estacionario. Adicionalmente, se recogen muestras a las 24h después de un cambio de dosis. Estas muestras se obtienen mediante punción venosa y se recogen en tubos de plasma que contienen heparina de litio (Vacuette S. A., Madrid). Seguidamente, son centrifugadas a 2.000g durante 10 min a 4°C y el sobrenadante obtenido es alicuotado y almacenado a -80°C hasta su procesamiento.
Resultados y discusiónCondiciones cromatográficasDurante el desarrollo del procedimiento de medida basado en la UHPLC-MS/MS se han combinado varias fases móviles y columnas específicas de UHPLC permitiendo optimizar la resolución y eficacia cromatográficas, así como obtener unos picos cromatográficos simétricos, unas relaciones S/N elevadas y unos tiempos de retención reducidos. En lo concerniente a las fases móviles evaluadas, la utilización de agua conjuntamente con acetonitrilo en lugar de metanol como solvente orgánico, ha dado lugar a una mejor separación cromatográfica y una mayor sensibilidad (metrológica) en el espectrómetro de masas. Por otra parte, el hecho de añadir ácido fórmico –en una proporción de volumen de 0,1%– a las fases móviles en lugar de acetato o formiato de amonio (a diferentes concentraciones) ha producido un incremento de la señal analítica en el detector como consecuencia de la disminución del ruido de fondo. Respecto a las columnas de UHPLC estudiadas (Acquity UPLC® BEH 2,1×50mm, 1,7μm; Acquity UPLC® BEH 2,1×100mm, 1,7μm; Acquity UPLC® HSS T3 2,1×50mm, 1,8μm y Acquity UPLC® HSS T3 2,1×100mm, 1,8μm; todas ellas de Waters), la Acquity UPLC® BEH 2,1×100mm, 1,7μm, en combinación con las fases móviles mencionadas y trabajando en la modalidad de gradiente, es la que nos ha permitido obtener una mejor resolución y eficacia cromatográficas.
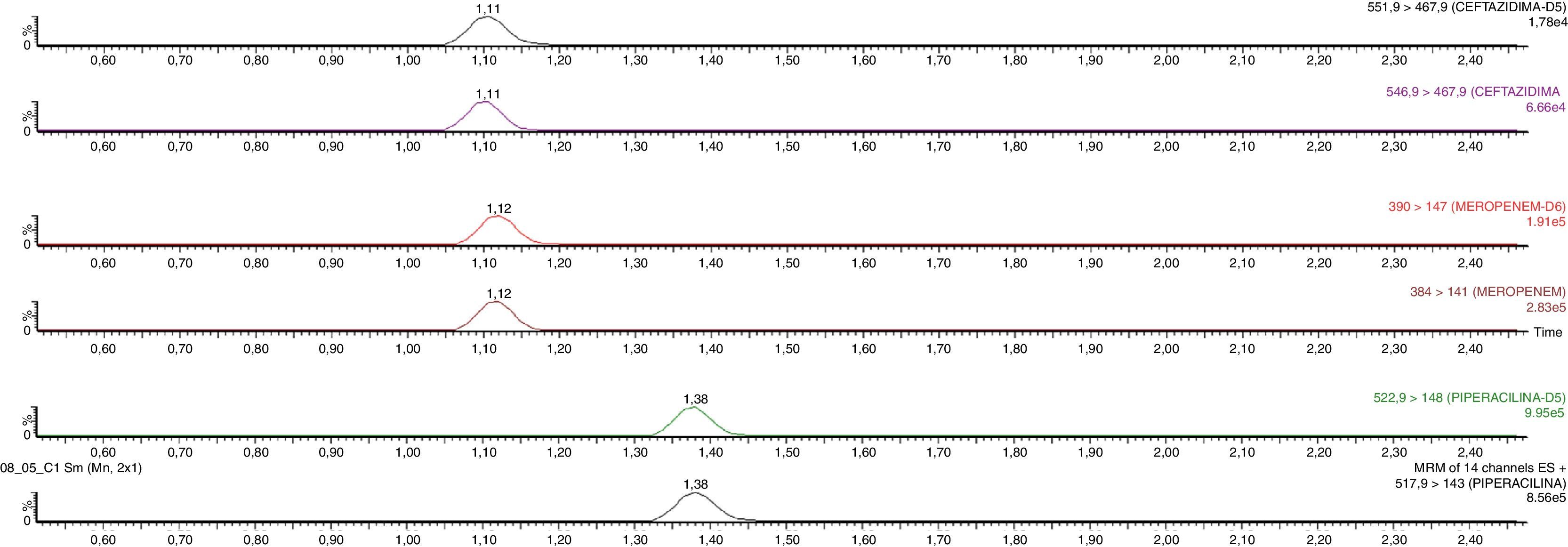
En las condiciones cromatográficas descritas, los tiempos de retención para la CFT, D5-CFT, MEM, D6-MEM, PIP y el D5-PIP son 1,11; 1,11; 1,12; 1,12; 1,38 y 1,38min, respectivamente (fig. 2). El tiempo de análisis es 3,5 min, inferior al obtenido en otros procedimientos previamente reportados9–11.
Condiciones del espectrómetro de masas
Los parámetros del espectrómetro de masas se han optimizado mediante la infusión directa de los antibióticos y sus PI, a una concentración de 10mg/l, en una solución de ácido fórmico 0,1% en agua: acetonitrilo (50:50 v/v) y a un flujo de 20μL/min. Para cada fármaco, la utilización de la ionización mediante electroespray en modo positivo (ESI+) ha permitido obtener una mayor sensibilidad metrológica, siendo los iones precursores más abundantes los correspondientes a los aductos del fármaco protonado ([M-H]+). La selección de los iones producto se ha llevado a cabo estudiando los patrones de fragmentación de los iones precursores en la célula de colisión del espectrómetro de masas en tándem. Todos los fármacos se han detectado trabajando en la modalidad MRM, monitorizando los iones precursores y los iones producto que han presentado una mayor señal analítica. Para evitar errores en la identificación de los antibióticos estudiados, así como para confirmar la ausencia de otros componentes similares en la muestra que hayan podido coeluir y contribuir falsamente la detección de estos fármacos, se han empleado dos transiciones MRM: una para la cuantificación y otra para la confirmación (tabla 1).
Preparación y tratamiento de las muestrasPese a que la precipitación de proteínas no es el procedimiento de preparación de muestras más idóneo para obtener recuperaciones aceptables y prevenir el efecto matriz que puede producirse durante el proceso de ionización de las muestras, en nuestro estudio, la combinación de una precipitación de proteínas con acetonitrilo seguida de una dilución subsiguiente del extracto con agua, simplifica el procedimiento de extracción publicado por otros autores4–16, y da lugar a valores de recuperación y efecto matriz aceptables (tabla 3).
ValidaciónProcedimiento de calibraciónTodas las curvas de calibración obtenidas son lineales entre 0,5mg/l y 175mg/l. A modo de ejemplo, alguna de las ecuaciones de calibración obtenidas son: y=2,6485·x+2,9698 (r2=0,9944) para CFT, y=0,1756·x+0,1759 (r2=0,9953 para MEM) y y=0,3947·x+0,1750 (r2=0,9995) para PIP. Las diferencias porcentuales relativas obtenidas, entre las concentraciones de masa de los antibióticos calculadas y teóricas, están comprendidas entre 1,5 y 15,0% para todos los materiales de calibración. Estos porcentajes cumplen con el criterio establecido por la EMA (± 15% y±20% a valores cercanos al LLOQ).
SelectividadDe acuerdo con la EMA13, un procedimiento de medida debe ser capaz de diferenciar los componentes de interés y sus PI de potenciales interferentes que puedan existir en la muestra (por ejemplo, fármacos concomitantes). A diferencia de otros estudios publicados9,10,12, se ha llevado a cabo un estudio de la selectividad procesando muestras de pacientes que están en tratamiento con otros fármacos. En nuestro caso, en ninguna de las muestras de pacientes procesadas se han obtenido picos interferentes en los tiempos de retención de la CFT, MEM, PIP y sus PI, indicando que el procedimiento de medida desarrollado presenta una adecuada selectividad.
Contaminación por arrastreLa guía de la EMA13 describe que, durante el desarrollo y validación de un procedimiento de medida, la contaminación por arrastre –si es que existe– debe eliminarse o, en su defecto, minimizarse. En contraste con la bibliografía publicada9–12, se ha realizado un estudio de la contaminación por arrastre. En nuestro caso, en los tres materiales de blanco procesados, no se han obtenido picos cromatográficos en los tiempos de retención de la CFT, MEM, PIP y sus PI, por lo que se puede descartar la existencia de una contaminación por arrastre.
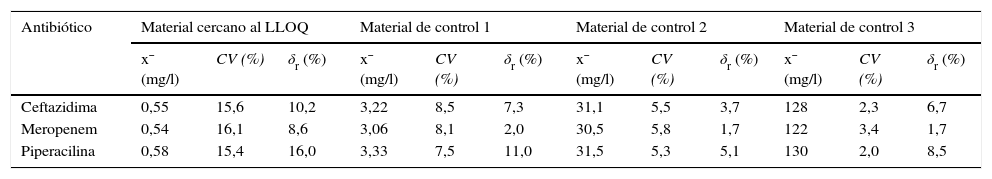
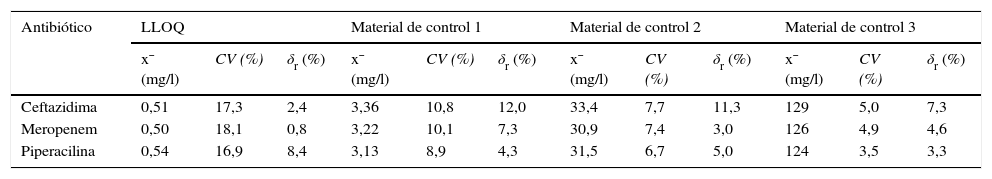
Límite de cuantificaciónLos LLOQ intraseriales obtenidos son 0,55mg/l (CV=15,6%; S/N=7,8) para la CFT, 0,54mg/l (CV=16,1%; S/N=6,9) para el MEM y 0,58mg/l (CV=15,4%; S/N=8,1) para la PIP; mientras que los LLOQ interdiarios son, para la CFT, el MEM y la PIP, 0,51mg/l (CV=17,3%; S/N=5,2), 0,50mg/l (CV=18,1%; S/N=5,5) y 0,54mg/l (CV=16,9%; S/N=5,6), respectivamente (tablas 4 y 5).
Valores de imprecisión y sesgo intraseriales obtenidos en el sistema de medida Acquity® UPLC®-TQD® para la concentración de masa de ceftazidima, meropenem y piperacilina en el plasma
| Antibiótico | Material cercano al LLOQ | Material de control 1 | Material de control 2 | Material de control 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x¯ (mg/l) | CV (%) | δr (%) | x¯ (mg/l) | CV (%) | δr (%) | x¯ (mg/l) | CV (%) | δr (%) | x¯ (mg/l) | CV (%) | δr (%) | |
| Ceftazidima | 0,55 | 15,6 | 10,2 | 3,22 | 8,5 | 7,3 | 31,1 | 5,5 | 3,7 | 128 | 2,3 | 6,7 |
| Meropenem | 0,54 | 16,1 | 8,6 | 3,06 | 8,1 | 2,0 | 30,5 | 5,8 | 1,7 | 122 | 3,4 | 1,7 |
| Piperacilina | 0,58 | 15,4 | 16,0 | 3,33 | 7,5 | 11,0 | 31,5 | 5,3 | 5,1 | 130 | 2,0 | 8,5 |
CV: coeficiente de variación; LLOQ: límite de cuantificación; δr: sesgo relativo; x¯: media.
Valores de imprecisión y sesgo interdiarios obtenidos en el sistema de medida Acquity® UPLC®-TQD® para la concentración de masa de ceftazidima, meropenem y piperacilina en el plasma
| Antibiótico | LLOQ | Material de control 1 | Material de control 2 | Material de control 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x¯ (mg/l) | CV (%) | δr (%) | x¯ (mg/l) | CV (%) | δr (%) | x¯ (mg/l) | CV (%) | δr (%) | x¯ (mg/l) | CV (%) | δr (%) | |
| Ceftazidima | 0,51 | 17,3 | 2,4 | 3,36 | 10,8 | 12,0 | 33,4 | 7,7 | 11,3 | 129 | 5,0 | 7,3 |
| Meropenem | 0,50 | 18,1 | 0,8 | 3,22 | 10,1 | 7,3 | 30,9 | 7,4 | 3,0 | 126 | 4,9 | 4,6 |
| Piperacilina | 0,54 | 16,9 | 8,4 | 3,13 | 8,9 | 4,3 | 31,5 | 6,7 | 5,0 | 124 | 3,5 | 3,3 |
LLOQ: límite de cuantificación; CV: coeficiente de variación; δr: sesgo relativo; x¯: media.
Estos resultados son similares a los descritos en la bibliografía8,9,12. En cambio, en otros estudios10,11, los LLOQ son inferiores a los obtenidos en el presente estudio, hecho que no es de extrañar si se tiene en cuenta que estos han empleado procedimientos de extracción más «limpios» (extracción en fase sólida o extracción líquido-líquido).
Imprecisión y sesgoLos valores de imprecisión y sesgo intraseriales e interdiarios obtenidos (tablas 4 y 5) no superan los requisitos metrológicos para la imprecisión y el sesgo relativo (en valor absoluto) establecidos por la EMA13 y el CLSI14,15 (≤ 15% y≤20% a valores cercanos al LLOQ), y son similares o inferiores a los obtenidos en publicaciones previas8–12. Los resultados obtenidos indican que el procedimiento de medida desarrollado es preciso y veraz.
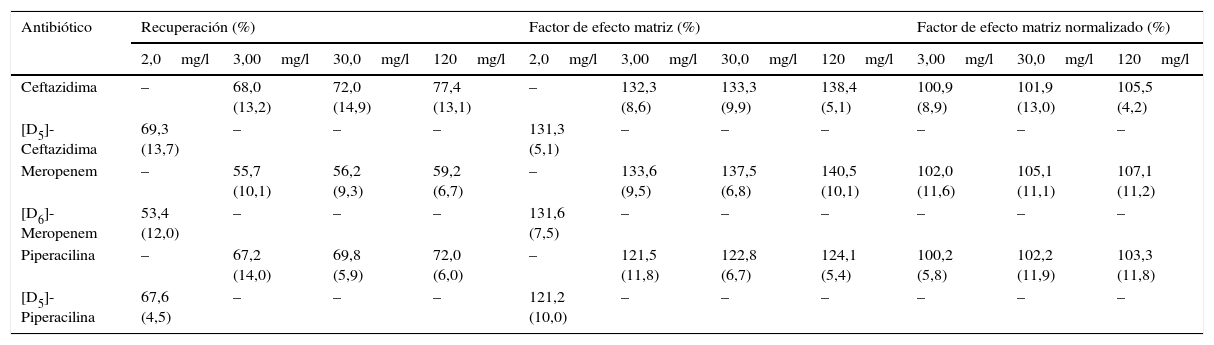
Eficiencia del proceso cromatográfico (recuperación y efecto matriz)Los valores de RE, ME y n-ME obtenidos se muestra en la tabla 3. Aunque se obtienen valores de RE relativamente bajos, la utilización de los PI seleccionados permite compensar la pérdida de los antibióticos durante el proceso de extracción de la muestra, con independencia de cuál sea el valor de la magnitud, y con una imprecisión aceptable (≤ 15%). En cuanto a los valores de ME, se puede observar que existe una sobreexpresión iónica para todos los antibióticos, que es compensada por la utilización de los PI. Los resultados obtenidos son similares a los publicados por otros autores8,9,12.
Valores de recuperación, factor de efecto matriz y factor de efecto matriz normalizado para la concentración de masa de ceftazidima, meropenem y piperacilina en el plasma
| Antibiótico | Recuperación (%) | Factor de efecto matriz (%) | Factor de efecto matriz normalizado (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2,0mg/l | 3,00mg/l | 30,0mg/l | 120mg/l | 2,0mg/l | 3,00mg/l | 30,0mg/l | 120mg/l | 3,00mg/l | 30,0mg/l | 120mg/l | |
| Ceftazidima | – | 68,0 (13,2) | 72,0 (14,9) | 77,4 (13,1) | – | 132,3 (8,6) | 133,3 (9,9) | 138,4 (5,1) | 100,9 (8,9) | 101,9 (13,0) | 105,5 (4,2) |
| [D5]-Ceftazidima | 69,3 (13,7) | – | – | – | 131,3 (5,1) | – | – | – | – | – | – |
| Meropenem | – | 55,7 (10,1) | 56,2 (9,3) | 59,2 (6,7) | – | 133,6 (9,5) | 137,5 (6,8) | 140,5 (10,1) | 102,0 (11,6) | 105,1 (11,1) | 107,1 (11,2) |
| [D6]-Meropenem | 53,4 (12,0) | – | – | – | 131,6 (7,5) | – | – | – | – | – | – |
| Piperacilina | – | 67,2 (14,0) | 69,8 (5,9) | 72,0 (6,0) | – | 121,5 (11,8) | 122,8 (6,7) | 124,1 (5,4) | 100,2 (5,8) | 102,2 (11,9) | 103,3 (11,8) |
| [D5]-Piperacilina | 67,6 (4,5) | – | – | – | 121,2 (10,0) | – | – | – | – | – | – |
En paréntesis se indican los coeficientes de variación (CV) obtenidos entre las diferentes muestras de pacientes.
Las diferentes magnitudes farmacológicas son estables 3 días a 5±3°C (PD ≤ −13,7%) y 6 meses a −80°C (PD ≤ −8,4%). Por otro lado, dentro del muestreador del sistema cromatográfico, estas son estables un máximo de 12 h a 4±1°C (PD ≤ −14,4%). Los valores negativos de las PD indican una descomposición o degradación de los antibióticos a lo largo del período de almacenamiento de las muestras. La reducida estabilidad de las magnitudes relacionadas con los antibióticos en estudio es un hecho conocido17–19, principalmente, en los extractos de las muestras y a temperatura ambiente. Por ello, es importante que los extractos se mantengan a temperatura de refrigeración durante un período de tiempo inferior a 12 h.
Aplicación clínicaLas concentraciones de masa de CFT, MEM y PIP en el plasma obtenidas para los pacientes críticos con sepsis se muestran en la tabla 2. A las 48 h del inicio del tratamiento, todos los valores fueron consistentes con la situación clínica de los pacientes (es decir, disminución o ausencia de fiebre, remisión de la infección, etc.) observada por los clínicos. Pese a ello, estos valores fueron claramente superiores a 4 veces la CMI del patógeno en cuestión y, en consecuencia, la dosis del antibiótico administrada fue reducida y mantenida hasta erradicar completamente la infección4–7. A las 72 h del inicio del tratamiento, los valores de la concentración de CFT, MEM y PIP disminuyeron y continuaron estando por encima de 4 veces la CMI (tabla 2).
ConclusionesEn este trabajo se ha desarrollado y validado un procedimiento de medida basado en la UHPLC-MS/MS que permite la medición simultánea de la concentración de masa de CFT, MEM y PIP en el plasma. Teniendo en cuenta el tiempo de análisis y las propiedades metrológicas obtenidas, este podría ser útil para la realización de estudios PK/PD y para la monitorización farmacoterapéutica de estos antibióticos en diferentes tipos de pacientes, particularmente, en pacientes críticos con sepsis, posibilitando así una mejor comprensión de la eficacia de estos fármacos en este tipo de pacientes.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
