


Las porfirias hepáticas agudas son 4 enfermedades raras causadas por deficiencias enzimáticas en la vía biosintética del grupo hemo. Se caracterizan por presentar síntomas neuroviscerales agudos potencialmente letales ante la presencia de factores inductores de la ALAS1. Estos factores pueden ser endógenos o exógenos tales como hormonas sexuales, ayuno, medicamentos, alcohol y tabaco, entre otros. La fisiopatología de los ataques involucra el incremento en la función de la ALAS1, la producción excesiva de precursores de porfirina y la alteración en la síntesis de hemoproteínas por la deficiencia relativa de hemo. En este artículo se revisa la interacción de esos mecanismos con algunos factores inductores, su papel en el origen de los síntomas neurológicos y cómo los tratamientos disponibles bloquean estos procesos.
The acute hepatic porphyrias are a group of 4 rare diseases caused by enzymatic deficiencies in the haem biosynthetic pathway. They are characterized by presenting acute attacks of neurovisceral symptoms in presence of factors that increase the ALAS1 activity. Those factors could be endogenous or exogenous, such as sexual hormones, fasting, drugs, alcohol, tobacco, among other. The physiopathology of the attacks involves an increasing in ALAS1 function, excessive production of porphyrin precursors, and disturbances in hemoproteins synthesis due to the relative haem deficiency. The present paper is a review of the interaction of those mechanisms with some factors that induce ALAS1, their role in the origin of neurovisceral symptoms, and how the available treatments interfere with those processes.
Las porfirias hepáticas agudas (PHA) son 4 enfermedades genéticas de muy baja prevalencia. Son causadas por mutaciones en los genes que codifican 4 enzimas de la vía metabólica del grupo hemo. La porfiria intermitente aguda (PIA) (OMIM: 176000), por deficiencia de hidroximetilbilano sintasa (HMBS); la coproporfiria hereditaria (OMIM: 121300), por deficiencia de coproporfirinógeno oxidasa (CPOX); y la porfiria variegata (OMIM: 176200), por deficiencia de protoporfirinógeno oxidasa (PPOX) son de herencia autosómica dominante con penetrancia incompleta. La plumboporfiria o porfiria de Doss (OMIM: 612740), por deficiencia de ácido aminolevulínico deshidratasa (ALAD) es la única de herencia autosómica recesiva. Aunque la síntesis de hemo está disminuida en los portadores de estas mutaciones, la cantidad producida es suficiente para cubrir sus necesidades basales, por lo que hasta el 90% de ellos no presentará síntomas de porfiria a lo largo de sus vidas1. Sin embargo, el grupo restante sufrirá de ataques agudos de síntomas neuroviscerales cuando los requerimientos de hemo aumenten por situaciones como ayuno prolongado o uso de medicamentos2. La fisiopatología de estos ataques aún no se ha esclarecido completamente, pero se sabe que la deficiencia de hemo, la ácido aminolevulínico sintasa 1 y el ácido aminolevulínico desempeñan un papel determinante. En este artículo se revisan diferentes mecanismos bioquímicos por los cuales estas moléculas, y su interacción con diferentes factores exógenos o endógenos, pueden ocasionar ataques agudos de porfiria, además de cómo los tratamientos disponibles contrarrestan esos eventos.
El grupo hemoEl hemo es el grupo prostético de las hemoproteínas, proteínas que se encargan de transportar y almacenar oxígeno, movilizar electrones y realizar reacciones de óxido-reducción. Algunas de estas son la hemoglobina, la mioglobina, los citocromos P450 (CYP450), la óxido nítrico sintasa, la triptófano pirrolasa, las catalasas y las peroxidasas, entre otras3.
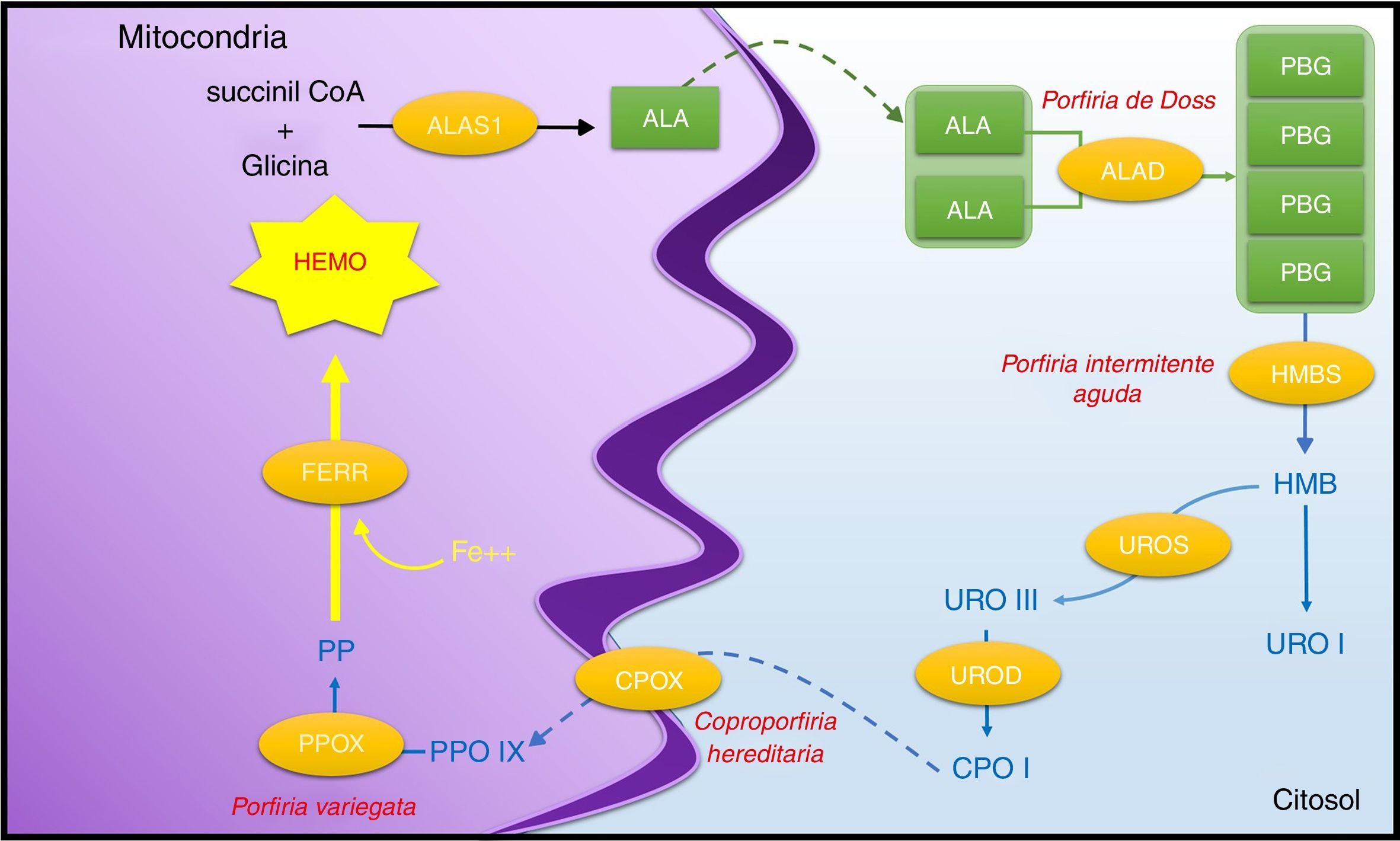
Biosíntesis del grupo hemoLa mayoría del hemo es sintetizado en la médula ósea y en el hígado; sin embargo, todas las células del organismo tienen dicha capacidad. En la médula ósea es sintetizado en las células eritroides en desarrollo, y más del 70% es utilizado para la producción de hemoglobina. En el hígado es elaborado en los hepatocitos y cerca del 65% se destina para generar CYP450. El proceso de biosíntesis involucra 8 reacciones enzimáticas, pero puede ser resumido en 5 grandes sucesos bioquímicos: la formación de un monopirrol, el ensamblaje de 4 monopirroles en un tetrapirrol lineal, la aromatización del tetrapirrol, las modificaciones secuenciales de los sustituyentes del anillo tetrapirrólico y la inserción del hierro a la protoporfirina ix. La formación del monopirrol se inicia en la mitocondria con la condensación de glicina y succinil CoA por medio de la ácido aminolevulínico sintasa (ALAS) para producir 5-ácido aminolevulínico (ALA). Posteriormente, 2 moléculas de ALA pasan al citosol, donde la ALAD las dimeriza para formar porfobirinógeno (PBG), que es el único monopirrol de la vía. Después, la HMBS ensambla 4 de esos monopirroles de PBG en un tetrapirrol lineal conocido como hidroximetilbilano. Subsecuentemente, este es ciclado a través de la uroporfobirinógeno sintasa (UROS) para formar un anillo tetrapirrólico conocido como uroporfobirinógeno iii (URO-III). A partir de este punto, todas las reacciones subsecuentes solo modifican los sustituyentes del anillo. En la primera modificación la uroporfobirinógeno descarboxilasa (UROD) produce coproporfobirinógeno iii (COPRO-III), el cual luego accede a la mitocondria donde la CPOX lo transforma en protoporfobirinógeno IX, que ulteriormente es convertido por la PPOX en protoporfirina ix, la única porfirina de la vía. Finalmente, la ferroquelatasa une el hierro con la protoporfirina ix y da origen al grupo hemo1–3 (fig. 1).

Vía biosintética del grupo hemo. Los precursores de porfirinas están en cajas verdes, las enzimas en óvalos amarillos.
ALA: ácido aminolevulínico; ALAD: ácido aminolevulínico deshidratasa; ALAS1: ácido aminolevulínico sintasa 1; CPO: coproporfirinógeno; CPOX: coproporfirinógeno oxidasa; FERR: ferroquelatasa; HMB: hidroximetilbilano; HMBS: hidroximetilbilano sintasa; PBG: porfobirinógeno; PP: protoporfirina; PPO: protoporfirinógeno; PPOX: protoporfirinógeno oxidasa; URO: uroporfobirinógeno; UROS: uroporfobirinógeno sintasa. El color de esta figura solo puede apreciarse en la versión electrónica del artículo.
En condiciones fisiológicas normales la síntesis de hemo es extremadamente eficiente y regulada; esto para garantizar que su concentración se encuentre siempre cercana a los requerimientos del cuerpo, ya que tanto el déficit como el exceso de este tienen un efecto citotóxico4. Su exceso es fuente de radicales libres de oxígeno que generan daño oxidativo de ácidos nucleicos, membranas lipídicas y proteínas. Además, puede causar anemia hemolítica al alterar la estabilidad de las membranas celulares y un estado proinflamatorio al reclutar leucocitos, plaquetas y glóbulos rojos al endotelio5. Su deficiencia perturba la formación de hemoproteínas con función energética y destoxificante, como las de la cadena respiratoria mitocondrial y los CYP450. La regulación de su metabolismo depende de la razón entre su concentración y las demandas fisiológicas, lo cual puede ser modificado por variaciones en las tasas de síntesis y degradación, controladas por la ALAS y la hemo oxigenasa-1 (HMOX1), respectivamente. No obstante, los mecanismos de regulación son diferentes en el hígado y en la médula ósea, debido a que la ALAS presenta isoformas específicas en estas localizaciones6. La ALAS1 se encuentra en todas las células no eritroides, incluidos los hepatocitos, y es codificada por genes en el cromosoma 3p21.2. La ALAS2 es específica de las células eritroides en desarrollo, y los genes que la codifican se localizan en el cromosoma Xp11.23.
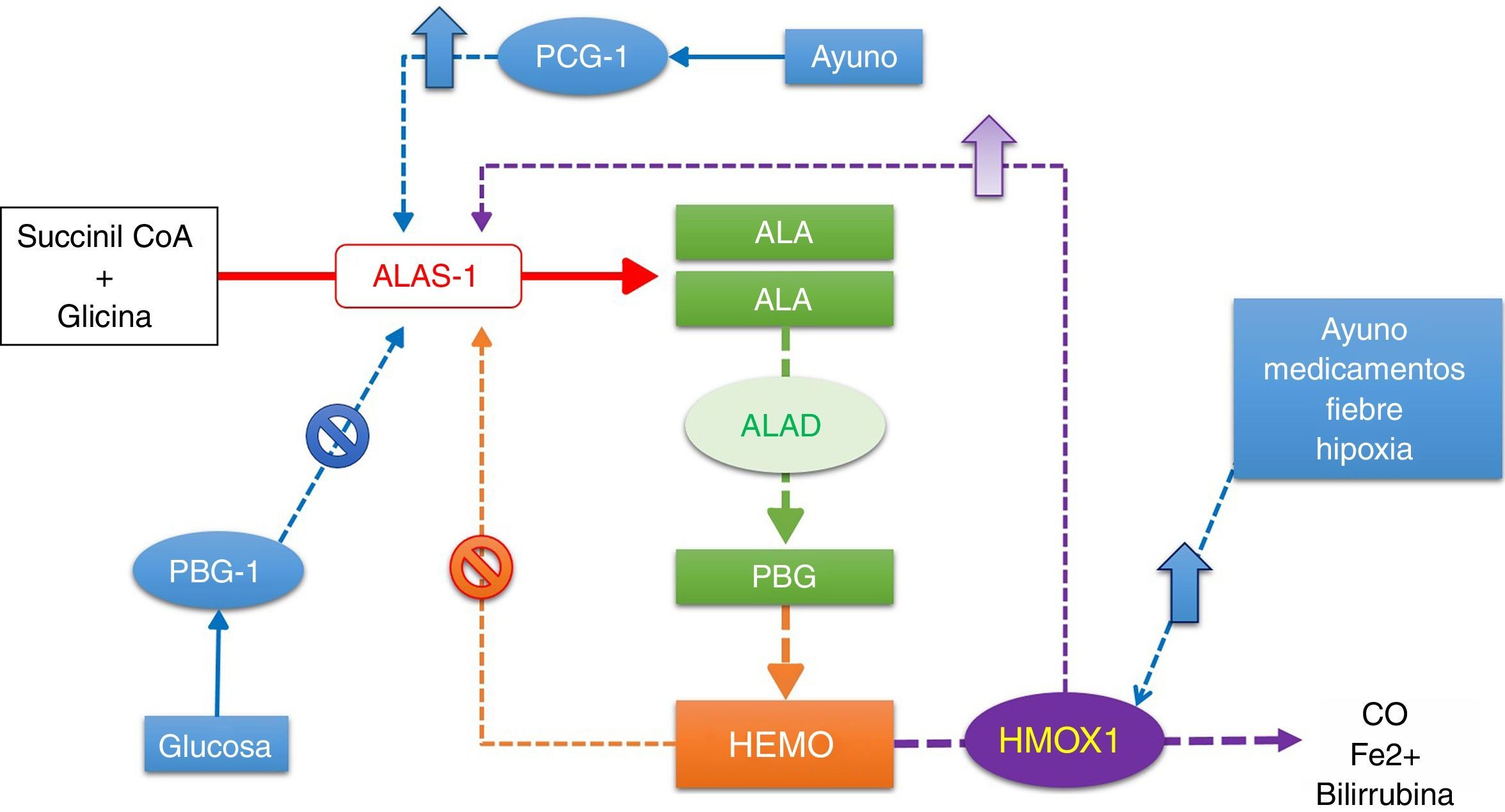
Ácido aminolevulínico sintasa 1La ALAS1 es la enzima limitante de la biosíntesis de hemo en hepatocitos y otras células no eritroides; esto por ser la de menor capacidad catalítica de la vía, por su rápido recambio (vida media entre 1-3h) y por ser fuertemente inducida cuando los requerimientos de hemo sobrepasan a la producción basal2. Por esta razón, la ALAS1 es regulada por numerosos y diversos mecanismos fisiológicos que modifican rápidamente la cantidad de esta enzima dependiendo del balance de hemo. Múltiples factores pueden actuar directa o indirectamente sobre estos mecanismos, activándolos o inactivándolos. Entre los más importantes se encuentran las concentraciones de hemo libre, glucosa, insulina y hormonas sexuales7,8. Sin embargo, todos los factores que bloquean la síntesis de hemo o aumentan su degradación tienen la capacidad de inducir a esta enzima (fig. 2).

Regulación de la síntesis de hemo en el hígado. Los factores que regulan la función de la ALAS1 regulan la síntesis de hemo. La ALAS 1 es inducida por la falta de hemo o el aumento en la demanda. La HMOX1 degrada el hemo. El ayuno, los medicamentos, la fiebre y la hipoxia inducen la HMOX1, y como consecuencia la ALAS1. El ayuno, además, induce a la ALAS1 por medio de los PCG-1. El hemo libre y la glucosa inhiben la ALAS1.
ALA: ácido aminolevulínico; ALAD: ácido aminolevulínico deshidratasa; ALAS1: ácido aminolevulínico sintasa 1; CO: dióxido de carbono; HMOX1: hemo oxigenasa 1; PBG: porfobirinógeno; PCG-1: receptores activados por proliferadores peroxisomales gamma coactivador transcripcional 1-alfa.
Esta isoenzima localizada en el retículo endoplasmático es la limitante de la degradación del hemo. Se encuentra en bajas cantidades en condiciones basales, pero es altamente inducible por estrés oxidativo y físico, ayuno, químicos, temperaturas elevadas (fiebre o hipertermia), hipoxia, isquemia-reperfusión, lipopolisacáridos y metales pesados5,9. La forma en la que estos factores actúan sobre la actividad de la HMOX1 es compleja y depende de múltiples interacciones con factores de transcripción como el Bach110,11, el factor nuclear derivado-eritroide-2 simil-2 (NFE2L2 o Nrf2)12 y la proteína activadora-1 (AP-1)13. Al ser inducida, la HMOX1 aumenta el catabolismo del hemo libre hepático reduciendo su concentración, lo que ocasiona un aumento en la actividad de la ALAS1.
Regulación de la ácido aminolevulínico sintasa 1 mediada por hemoEl grupo hemo regula su propio metabolismo al inhibir por contrarregulación a la ALAS1. Aunque no tiene un efecto directo sobre el centro catalítico de la enzima1, modifica su síntesis a nivel transcripcional14 y traduccional15, impide su importación desde el citosol a la mitocondria16 y aumenta su degradación17. Los mecanismos involucrados en estos procesos aún no han sido completamente dilucidados. Una de las hipótesis plantea que el principal mecanismo regulador serían las variaciones rápidas en la concentración de hemo libre en diversos depósitos intracelulares. Este hemo libre es una pequeña cantidad que es almacenada antes de formar parte de las hemoproteínas como grupo prostético. Sus depósitos se encuentran en la mitocondria, el retículo endoplasmático y el citosol, y en cada localización regula de forma diferente su propio metabolismo2. En la mitocondria controlan la tasa de síntesis de la citocromo oxidasa, en el retículo endoplasmático la actividad de la hemo oxigenasa y en el citosol la síntesis de la ALAS1. A este último se le conoce como hemo regulador debido a que modula directamente la enzima limitante de la vía2. Anteriormente, se creía que su efecto regulador se debía a que impedía la transcripción de ARN mensajero de la ALAS1 (ARNm-ALAS1), pero se ha demostrado que más que impedir la transcripción lo que hace es alterar la estabilidad del ARNm-ALAS1 por medio de modificaciones postranscripcionales, lo que reduce su vida media y consecuentemente la producción de ALAS118.
Regulación de la ácido aminolevulínico sintasa 1 mediada por glucosa e insulinaLas concentraciones de glucosa e insulina regulan indirectamente a la ALAS1 por medio de su efecto sobre los receptores activados por proliferadores peroxisomales gamma coactivador transcripcional 1-alfa (PGC-1α). Estos regulan positivamente la transcripción del gen ALAS1 (OMIM: 125290) al coactivar al NFR1 (factor-1 respiratorio nuclear) y al FOXO1 (forkhead box O1), los cuales están unidos directamente al promotor de la ALAS17. La insulina disminuye y el glucagón aumenta la expresión de PGC-1α. Por esta razón la glucemia, al modular tanto los niveles de insulina como de glucagón, controla este mecanismo de regulación de la ALAS1. La insulina modula indirectamente la función de los PGC-1α al activar la proteína quinasa Akt en el hígado, la cual fosforila al FOXO17. Cuando FOXO1 esta fosforilado se separa de los PGC-1α y es exportado del núcleo al citosol, impidiendo la acción transcripcional de los PGC-1α sobre gen ALAS1, y de esa forma suprime la síntesis enzimática19,20. El efecto represor aislado de la insulina sobre la ALAS1 es mayor que el de la glucemia, pero el efecto conjunto de ambas es mucho más alto que el de la insulina sola7. La hipoglucemia causa un aumento en los niveles de glucagón y la sobreexpresión de los PGC-1α y aumenta la actividad de la ALAS121. El ayuno induce a la ALAS1 por medio de la hipoglucemia que le caracteriza, pero además por aumentar la degradación de hemo por medio de la hemo oxigenasa-1 (HMOX1)22. Por otro lado, el hemo y la ALAS1 tienen un patrón de variación circadiano que es mediado en parte por su interacción con los PGC-1α y el receptor nuclear Rev-erbα23. Cuando el hemo se une al Rev-erbα recluta al complejo correpresor NCoR/histona desacetilasa 3 (HDAC3), el cual tiene la capacidad de reprimir la transcripción del PGC-1α. De esta forma, el Rev-erbα regula negativamente la homeostasis de hemo, cuando sus niveles son bajos se inducen los PGC-1α y aumenta la transcripción de ALAS123.
Regulación de la ácido aminolevulínico sintasa 1 mediada por factores hormonalesLas hormonas sexuales se encuentran entre los más importantes factores reguladores de la ALAS1. Evidencia de esto es que los AAP son infrecuentes antes de la pubertad, que las mujeres en edad fértil son más sintomáticas que los hombres, que en ellas los AAP se inician más tempranamente, que algunas pueden sufrir de AAP cíclicos durante la fase lútea de la menstruación (cuando los niveles de progesterona son más altos), que estos ataques premenstruales pueden ser prevenidos con la administración de análogos de hormona liberadora de gonadotropina (GnRH), que la frecuencia de AAP disminuye después de la menopausia y que el uso de anticonceptivos orales puede desencadenar AAP24,25. La progesterona y los estrógenos son metabolizados en el hígado por los CYP450, lo cual aumenta las demandas de hemo para la formación de dichas hemoproteínas y explica la capacidad de estas hormonas para inducir la ALAS125. Adicionalmente, la progesterona aumenta el catabolismo del hemo al inducir la HMOX1, por lo que se le considera la principal hormona relacionada con los AAP24.
Teniendo en cuenta este efecto de las hormonas sexuales sobre la ALAS1, sería esperable que durante la gestación y el puerperio incrementaran la frecuencia y gravedad de los AAP a la par con el marcado incremento de los niveles hormonales. Sin embargo, la mayoría de las mujeres con PHA toleran bien estos estados y solo algunas presentan crisis26. Kauppinen et al. reportaron que hasta el 92% de las mujeres gestantes con PHA no sufren de AAP26, lo cual ha sido corroborado por otros investigadores27,28. Por lo tanto, la gestación no está contraindicada en estas mujeres, siempre que se les monitorice regularmente27. Este efecto paradójico de la gestación sobre los AAP se ha explicado por cambios en el metabolismo de la progesterona, que sobrepasan la capacidad de la hormona para inducir a la ALAS1 y que como resultado neto protegen a la mujer gestante de los AAP2. Cuando suceden, los AAP gestacionales usualmente se deben al uso de medicamentos (como metoclopramida para la hiperémesis gravídica) más que a la gestación como tal25.
Regulación de la ácido aminolevulínico sintasa 1 mediada por medicamentosMuchos medicamentos tienen la capacidad de inducir a la ALAS1, incluso a bajas dosis. Entre los mecanismos para ello se encuentran: la inducción de los CYP450, la inducción de la ALAS1 por medio de factores de transcripción, el agotamiento de los depósitos de hemo libre y la degradación directa del grupo hemo2,29. Los medicamentos pueden inducir la expresión de CYP450 por medio de varios receptores nucleares. Algunos de estos también tienen la capacidad de aumentar la transcripción de ARNm-ALAS1, debido a que la región 5’ del gen ALAS1 posee varios elementos de respuesta transcripcional a medicamentos (ADRES) que son activados mediante receptores huérfanos que actúan como factores de transcripción como el Chicken Xenobiotic-sensing Receptor30, Pregnane X Receptor31 y Constitutive Androstane Receptor que responden a medicamentos y xenobióticos, e inducen la transcripción de la ALAS132. Igualmente, el gen ALAS1 tiene una región de respuesta altamente conservada para el receptor nuclear farsenoide X (FXR) activado por ácidos biliares. Este receptor nuclear es importante para conservar la homeostasis de los ácidos biliares al reprimir sus síntesis de novo y aumentar su degradación por medio de la sobreexpresión de CYP45033. Por medio de este mecanismo, los ácidos biliares naturales o sintéticos, al inducir a la ALAS1 y los CYP podrían precipitar AAP en ciertos pacientes, como aquellos con enfermedad colestásica33. En resumen, la ALAS1 en respuesta al uso de medicamentos es mediada por los mismos mecanismos de control de expresión de los CYP450 y suceden coordinadamente32. En un segundo momento, la inducción de ALAS1 también se debe a que los depósitos de hemo libre se agotan en la producción de CYP450 para el metabolismo de los medicamentos13,32.
Numerosos principios activos han sido identificados y clasificados como seguros o inseguros en pacientes con PHA. Sin embargo, para muchos otros el conocimiento es aún escaso, dado que no es posible clasificarlos en función de los estudios de laboratorio, pues la respuesta a estos difiere entre pacientes, e incluso en un mismo paciente en diferentes momentos de su vida34. Por ello, se han desarrollado algoritmos predictivos sustentados en las propiedades químicas y farmacológicas de los principios activos para definir y clasificar la seguridad del uso de ciertos medicamentos en estos pacientes29. Teniendo esto en cuenta, los medicamentos son asignados a una de 6 categorías (no porfirinogénico, probablemente no porfirinogénico, posiblemente porfirinogénico, probablemente porfirinogénico, porfirinogénico y no clasificado aún)29. Siempre que sea posible deben utilizarse los medicamentos que estén clasificados como no porfirinogénicos. Para conocer la clasificación de los medicamentos pueden consultarse múltiples bases de datos en línea que se actualizan constantemente como: www.drugs-porphyrias.org – www.porphyiasfoundation.com.
Regulación de la ácido aminolevulínico sintasa 1 mediada por alcohol y tabacoEl alcohol inhibe numerosas enzimas relacionadas con la biosíntesis de hemo, induce la ALAS1 y algunos CYP450. En pacientes con PHA asintomáticas el alcoholismo crónico y el consumo agudo>80g de alcohol pueden causar un aumento en la producción de porfirinas y sus precursores sin desencadenar AAP, lo que se conoce como descompensación latente35. En este sentido, consumos>60g/día pueden aumentar hasta 6 veces los niveles de ALA, PBG y porfirinas excretadas36. Los pacientes con PHA y dependencia al alcohol pueden permanecer con niveles basales elevados de porfirinas, lo cual les confiere un mayor riesgo de sufrir de AAP36. Este mismo efecto lo tienen los alcoholes no etílicos, como los polifenoles presentes en el vino tinto o la combinación de alcoholes en otras bebidas37. Los mecanismos por los cuales el alcohol induce a la ALAS1 no han sido elucidados. Estos pueden estar en relación con que el alcohol ocasiona un agotamiento de los depósitos de hemo libre para la producción de CYP450, aumenta la absorción de ciertos medicamentos inductores de la ALAS1 (como fenobarbital), reduce el metabolismo de esos medicamentos y con los metabolitos del metabolismo alcohólico (acetaldehído o acetato)35. Por ello, los pacientes polimedicados que son alcohólicos son los que tienen un mayor riesgo de AAP. Por otro lado, el cigarrillo se asocia con AAP recurrentes por estar compuesto de múltiples químicos que alteran el metabolismo de algunos esteroides sexuales, inducen a los CYP450 y la ALAS138. Por esa razón dejar de fumar podría ser particularmente beneficioso en el control de los AAP recurrentes.
Ataques agudos de porfiriaSe conoce como AAP al desarrollo rápido de síntomas neuroviscerales en pacientes con PHA cuando se exponen a factores inductores de la ALAS1 (endógenos o exógenos)39. Estos ataques son muy heterogéneos y abarcan una gran cantidad de posibles manifestaciones clínicas que son indistinguibles entre las diferentes PHA40. Durante estos episodios la actividad de la ALAS1 supera la función normal de la ALAS2 en la médula ósea y bajo esas condiciones la HMBS, con una actividad catalítica cercana a la de la ALAS1 en condiciones basales, se convierte en la enzima limitante de la producción de hemo. Por esa razón, los metabolitos producidos en los pasos anteriores o precursores de porfirinas (ALA y PBG) se acumulan en cantidades excesivas. Esto ocurre en todas las PHA, pero es más marcada en la PIA por la actividad disminuida de la HMBS1.
Fisiopatología de los ataques agudos de porfiriaEl mecanismo exacto por el cual suceden las alteraciones neurológicas de los AAP es desconocido. Se ha postulado que estos se deben a que los precursores de porfirinas (ALA y PBG) son neurotóxicos y a que la deficiencia de hemo causa una alteración en la formación y funcionamiento de las hemoproteínas1.
Neurotoxicidad de los precursores de porfirinasEl ALA y el PBG tradicionalmente han sido considerados los principales responsables de los síntomas neurológicos de los AAP. Sin embargo, recientemente el papel del PBG fue desestimado por varios estudios y el ALA pasó a ser el principal implicado. En un estudio se suministró PBG desaminasa recombinante humana (Porphozyme®) a pacientes con niveles elevados de ALA y PBG, con lo cual se consiguió un descenso rápido de los niveles de PBG, pero sin modificar los niveles de ALA ni los síntomas41. Esto contrasta con lo que pasa en pacientes con PHA que reciben trasplantes hepáticos, en quienes tanto los niveles de ALA como de PBG se normalizan y los síntomas desaparecen42. Algo similar ha sido observado en pacientes con tirosinemia hereditaria tipo i o con intoxicación por plomo. Estos presentan síntomas neurológicos que simulan a los AAP y niveles elevados de ALA, pero sin incremento del PBG, y luego de suministrarles hemina intravenosa (sin ser este el tratamiento) los niveles de ALA se normalizan y los síntomas desaparecen43. El efecto neurotóxico del ALA ha sido demostrado en modelos experimentales con células nerviosas44,45. Este guarda relación con su capacidad de promover la producción de radicales libres de oxígeno y causar daño oxidativo del ADN nuclear y mitocondrial46,47. Además, se ha postulado que su neurotoxicidad también podría estar relacionada con su habilidad para ingresar en las células nerviosas, donde se convierte en porfirinas con efecto citotóxico1, y a que por el gran parecido estructural con el ácido gamma amino butírico puede activar sus receptores y suprimir el sistema nervioso2,48,49. Sin embargo, los niveles elevados de ALA no son suficientes para causar un AAP, como fue demostrado por un estudio en el que se administró ALA a pacientes sanos en cantidades similares a las vistas durante un AAP sin lograr desencadenar síntomas neuroviscerales50. Así mismo, se ha observado que los niveles de ALA no se correlacionan con la gravedad clínica de los AAP1,51.
Formación anómala de hemoproteínasEl hemo es un componente esencial de muchas proteínas y enzimas, por lo que su deficiencia puede afectar la formación y función de estas. La alteración en algunas de estas hemoproteínas puede ser la explicación a muchos de los síntomas observados durante los AAP. Por ejemplo, el mal funcionamiento de las proteínas de la cadena transportadora de electrones en la mitocondria induce un déficit de energía que afecta la función neural, el transporte axonal52 y causa muerte neuronal53,54. La falta de ATP altera la función de la Na+/K+ ATPasa, la cual aporta 15mv del potencial de membrana neuronal en condiciones normales, por lo que su mal funcionamiento ocasiona una alteración en la excitabilidad de la membrana axonal que se normaliza después del tratamiento55,56. La óxido nítrico sintasa no logra producir suficiente óxido nítrico, y como consecuencia se deteriora el flujo sanguíneo cerebral y esplácnico57–59. La disfunción de la triptófano pirrolasa trastorna el metabolismo de la serotonina, lo cual podría explicar algunos de los síntomas nerviosos centrales60. La alteración de la función de los CYP450 y otras enzimas antioxidantes disminuye la capacidad destoxificante del organismo y ocasiona la acumulación de especies reactivas de oxígeno que lesionan las membranas celulares y el ADN43.
Tratamiento de los ataques agudos de porfiriaSupresión de la ácido aminolevulínico sintasa 1La administración de hidratos de carbono (dextrosa) y hemina intravenosa son consideradas las terapias específicas para los AAP, debido a su habilidad para suprimir la ALAS1 y reducir la producción de ALA y PBG. En algunos casos se ha usado cimetidina con buenos resultados, pero la evidencia es muy escasa. Ninguna de estas medidas revierte el daño neurológico, por eso los pacientes con AAP avanzados pueden permanecer sintomáticos a pesar del tratamiento. Los síntomas desaparecen paulatinamente en tanto las fibras nerviosas se regeneran fisiológicamente61. El efecto represor de los hidratos de carbono sobre la ALAS1 es explicado por su interacción con los PGC-1α7. Son el tratamiento estándar para los ataques leves, pero deben ser administrados a todos los pacientes sin importar la gravedad clínica para prevenir episodios inadvertidos de hipoglucemias ocasionados por ayuno. Se debe garantizar un aporte de 300-500g/día, que pueden ser administrados por vía oral o sublingual, por medio de bebidas azucaradas, o intravenosa en forma de dextrosa62. Debe tenerse en cuenta que la respuesta clínica a los hidratos de carbono no es igual en todos los pacientes, pues algunos responden inmediatamente y otros no lo hacen14. Por otro lado, la hemina intravenosa cuando entra al espacio vascular se une a las proteínas plasmáticas, albúmina y hemopexina, y es captada primariamente por los hepatocitos donde repone los depósitos de hemo libre, y de esa forma suprime a la ALAS1. Es la medida terapéutica disponible más efectiva, con un demostrado impacto en disminuir el dolor, la producción de precursores de porfirinas y la estancia hospitalaria63,64. Está indicada solo para los AAP graves. Finalmente, la cimetidina puede suprimir la ALAS1 indirectamente al inhibir varios CYP450 y la hemo oxigenasa microsómica, lo que disminuye la degradación del hemo65.
Terapias emergentesAunque la hemina intravenosa es la mejor terapia disponible, y ha demostrado ser efectiva en revertir los AAP, tiene ciertas limitaciones que requieren ser superadas con nuevos tratamientos, como su inicio de acción lento, la necesidad de múltiples infusiones diarias (típicamente más de 3) para normalizar los niveles elevados de ALA y PBG y el riesgo de efectos adversos graves como flebitis, anafilaxia y sobrecarga de hierro63. Recientemente, se evaluó una nueva terapia con ARN interferente pequeño dirigido al hígado por nanopartículas lipídicas para silenciar directamente la expresión del gen ALAS1 (ALAS1-ARNip). En este estudio la terapia con ALAS1-ARNip fue más efectiva que la hemina en reducir los niveles de ALA-PBG y tratar los AAP en modelos murinos de PIA a los que se les suministró fenobarbital. Igualmente, el efecto terapéutico se logró más rápidamente, y con la administración profiláctica se logró un efecto preventivo prolongado. Su uso no afectó la función hepática, los niveles de hemo almacenado ni la función de las hemoproteínas, que son preocupaciones que surgen al inhibir fuertemente la función de la ALAS166. Por otro lado, otro estudio demostró la seguridad de la terapia génica con vectores adenovirales codificantes de PBGD (rAAV5-cohPBGD) en primates no humanos67. Posteriormente, esta terapia fue evaluada en un ensayo clínico de fase i donde se evidenció que su uso es seguro en humanos, pero inefectivo en reducir los niveles de ALA y PBG68.
ConclusionesEsclarecer los mecanismos fisiopatológicos de los AAP es importante para su prevención y tratamiento. En este sentido, los principales implicados son la inducción excesiva de ALAS1, la neurotoxicidad del ALA y la deficiencia de hemo. Los AAP son desencadenados por factores endógenos y exógenos con la capacidad de inducir la enzima ALAS1, directamente o indirectamente. La mayoría de los mecanismos indirectos guardan relación con alteraciones en la homeostasis del hemo y la habilidad de diversas sustancias para aumentar sus demandas o degradación, esto porque el hemo libre regula su propia tasa de síntesis con base en su concentración, por medio de mecanismos postranscripcionales que alteran la estabilidad del ARNm-ALAS1. Factores como el estrés, el ayuno, la fiebre, la hipoxia, la progesterona y algunos químicos aumentan por diferentes vías la actividad de la HMOX1 y la degradación de hemo. Algunos medicamentos, los estrógenos, el alcohol y las sustancias que se encuentran en el humo de los cigarrillos inducen los CYP450 y aumentan las demandas de hemo para la síntesis de estas hemoproteínas. El ayuno causa la sobreexpresión de los receptores PGC-1α, que regulan positivamente la actividad de la ALAS1 y aumentan su expresión. La inducción exagerada de la ALAS1 ocasiona un aumento equiparable en la producción de ALA. El origen de los síntomas se debe en gran parte al efecto neurotóxico de este precursor de porfirinas por su potencial oxidativo y de lesionar el ADN y a la alteración de la función de las hemoproteínas por la deficiencia de hemo durante los AAP. Basándonos en los mecanismos fisiopatológicos la terapia para los AAP consiste en suprimir la ALAS1. La terapia con glucosa se vale de suprimir los receptores PGC-1α para inhibir indirectamente la ALAS1 y la terapia con hemina de los mecanismos de autorregulación del hemo libre al repletar sus depósitos. No obstante, estas terapias tienen problemas que deben ser superados, como su lenta acción y la presencia de efectos adversos graves, por lo que se deben explorar nuevas alternativas terapéuticas. Actualmente se están conduciendo estudios para evaluar la utilidad clínica del ALAS1-ARNip y la terapia génica con vectores adenovirales, y aunque los resultados son alentadores aún se requiere más investigación para comprobar su utilidad en humanos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.
A la señora Daniela Carrasquilla Zuluaga por sus invaluables aportes en la redacción y corrección del texto.
