


La homocistinuria es un error innato del metabolismo de la metionina con una alta tasa de morbimortalidad. Mutaciones en la cistationina beta-sintetasa son la causa más frecuente de homocistinuria, conocida esta como homocistinuria clásica. Hay descritas más de 150 mutaciones diferentes, de las cuales la más prevalente en España es la T191M. La detección mediante cribado neonatal puede prevenir las complicaciones más graves de la enfermedad y posibilitar un desarrollo cognitivo normal. Presentamos un caso de homocistinuria clásica debida a la mutación T353M, con fenotipo no respondedor a piridoxina.
Homocystinuria is an inherited disorder of methionine metabolism, and has a high morbidity-mortality rate. Mutations in the cystathionine beta-synthase gene are the most common cause of homocystinuria, known as classic homocystinuria. More than 150 mutations have been described, with T191M being the most prevalent in Spain. Neonatal identification by newborn screening may prevent severe complications, and allow normal intellectual development. A case is presented of pyridoxine non-responsive homocystinuria due to T353 mutation.
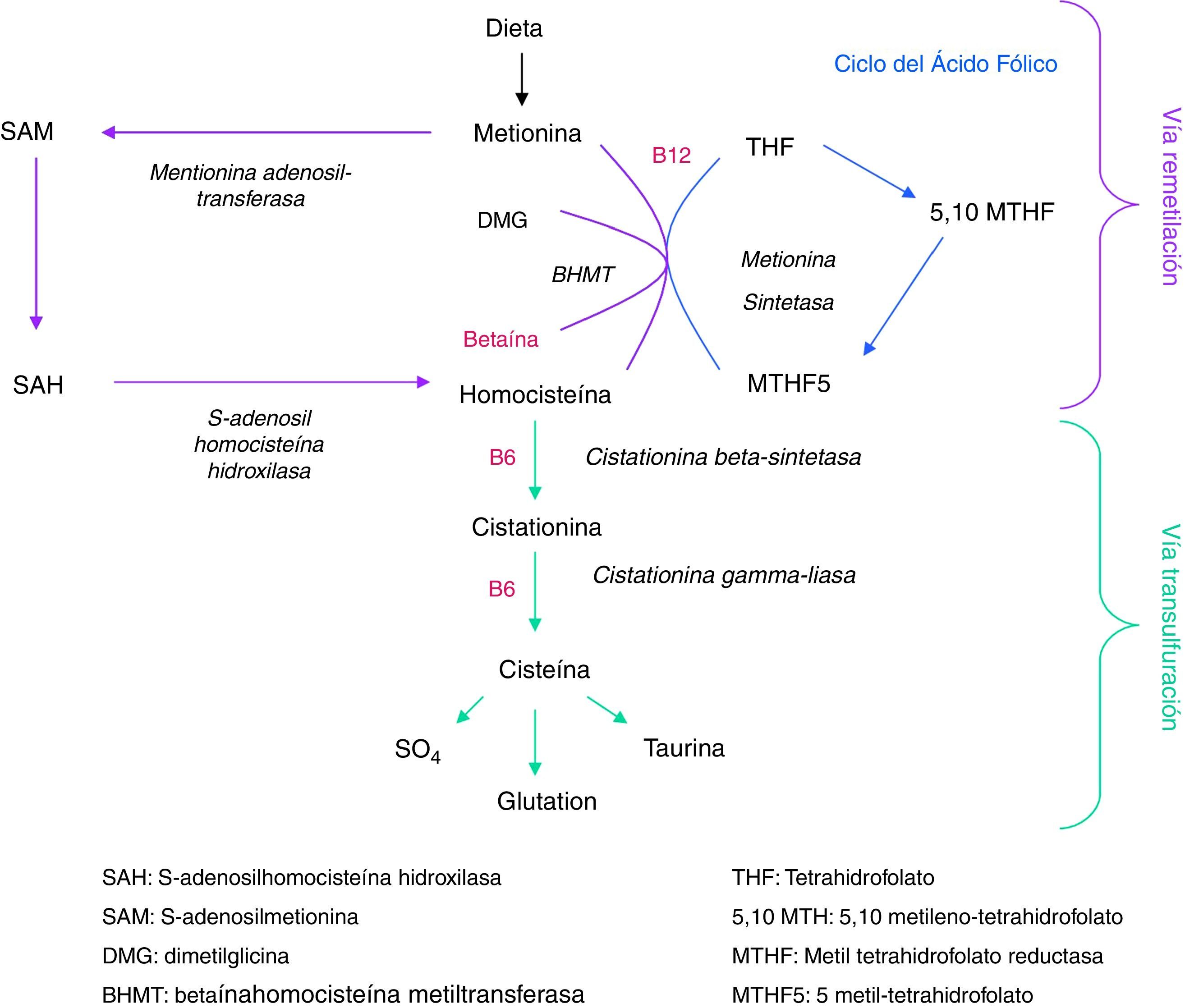
La homocistinuria se debe a un error congénito del metabolismo que provoca un acúmulo de homocisteína en plasma, orina y tejidos1. La hiperhomocisteinemia se puede producir por un error congénito del metabolismo o por trastornos adquiridos (tabla 1)2,3. La metionina, procedente de la dieta o del catabolismo de las proteínas endógenas, es transformada a homocisteína. La homocisteína puede metabolizarse por 2vías diferentes: transulfuración y remetilación (fig. 1).
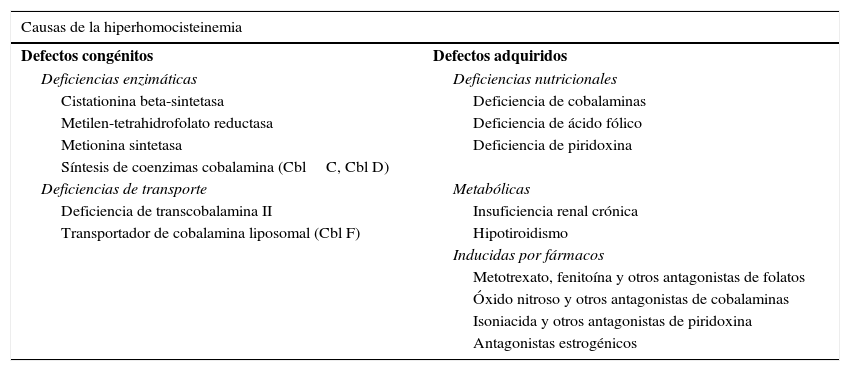
Causas de la hiperhomocisteinemia
| Causas de la hiperhomocisteinemia | |
|---|---|
| Defectos congénitos | Defectos adquiridos |
| Deficiencias enzimáticas | Deficiencias nutricionales |
| Cistationina beta-sintetasa | Deficiencia de cobalaminas |
| Metilen-tetrahidrofolato reductasa | Deficiencia de ácido fólico |
| Metionina sintetasa | Deficiencia de piridoxina |
| Síntesis de coenzimas cobalamina (CblC, Cbl D) | |
| Deficiencias de transporte | Metabólicas |
| Deficiencia de transcobalamina II | Insuficiencia renal crónica |
| Transportador de cobalamina liposomal (Cbl F) | Hipotiroidismo |
| Inducidas por fármacos | |
| Metotrexato, fenitoína y otros antagonistas de folatos | |
| Óxido nitroso y otros antagonistas de cobalaminas | |
| Isoniacida y otros antagonistas de piridoxina | |
| Antagonistas estrogénicos | |
Fuente: Tomada de Aguirre Errastia y Egurbide Arberas2.

Mediante la transulfuración, la homocisteína se transforma a cisteína por medio de 2reacciones dependientes de la vitamina B6. La primera de ellas es catalizada por la cistationina beta-sintetasa y, en ella, la homocisteína se condensa con una molécula de serina para formar cistationina. En la segunda reacción, la cistationina es transformada a cisteína por acción de la cistationina-gamma-liasa. La cisteína es finalmente catabolizada, y se elimina por la orina en forma de sulfato4.
En la ruta de la remetilación, la homocisteína se metila para formar metionina mediante 2rutas metabólicas independientes, en las que participan respectivamente las enzimas 5-metiltetrahidrofolato-reductasa y la betaína-homocisteína-metiltransferasa5. La primera de estas enzimas requiere 5-metiltetrahidrofolato y metilcobalamina como fuentes de grupos metilo. La segunda emplea betaína como fuente de grupos metilo. Por tanto, las vitaminas del grupo B (piridoxina, ácido fólico y metilcobalamina) son necesarias en el metabolismo de la metionina y la homocisteína, y tienen un importante papel terapéutico en los pacientes afectos de homocistinuria.
La deficiencia de cistationina beta-sintetasa es la más frecuente, por lo que se denomina homocistinuria clásica. Posee un patrón de herencia autosómica recesivo5.
Se han descrito gran número de mutaciones asociadas con la enfermedad. Las de mayor prevalencia son: I278T, que responde a piridoxina (vitamina B6), y G307S, que no responde a B61,6. No obstante, en España, la mutación T191M, sin respuesta a piridoxina, es la más prevalente7–9.
El acúmulo de homocisteína en los tejidos es tóxico, afecta particularmente a los sistemas óseo, ocular, nervioso y vascular, y el principal motivo de morbimortalidad son los fenómenos tromboembólicos4,10–12.
El tratamiento consiste en aumentar la actividad enzimática residual, con dieta baja en metionina y en proteínas totales, y administrar betaína en pacientes no respondedores a la piridoxina. Su objetivo es mantener los niveles de homocisteína dentro del rango de normalidad o lo más cercano posible. Se considera un control metabólico aceptable niveles <50μmol/L4.
La respuesta a piridoxina está influida por los valores de folato, por lo que se debe aportar ácido fólico y se ha de monitorizar la vitamina B12, y suplementarla en caso de déficit. Para los no respondedores se incluye la betaína, que favorece la remetilación de homocisteína, con lo que descienden los niveles de esta10. La betaína puede incrementar las concentraciones de metionina y debe monitorizarse, ya que se han descrito 3 casos de edema cerebral asociado si los valores en plasma exceden 1.000μmol/L13.
Un desarrollo cognitivo normal y la prevención de complicaciones físicas es posible en pacientes detectados mediante cribado neonatal, tratados de manera precoz y con un estricto cumplimiento11.
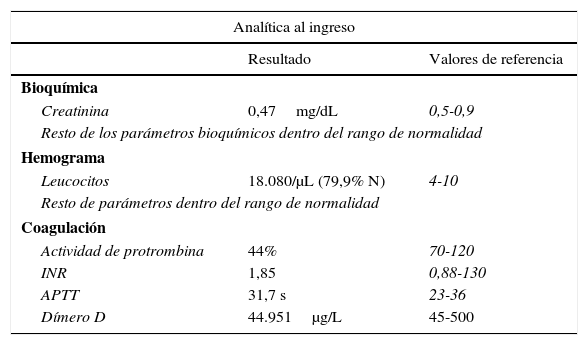
Presentación del casoMujer de 14 años que fue derivada a urgencias por edema y dolor de miembros inferiores. El juicio clínico fue trombosis venosa profunda con afectación de todo el eje íleo femoropoplíteo izquierdo y trombosis superficial de la vena safena interna, junto con un síndrome de Cockett. Se ingresó para la realización de fibrinólisis. Los resultados analíticos de la paciente se muestran en la tabla 2.
Resultados analíticos
| Analítica al ingreso | ||
|---|---|---|
| Resultado | Valores de referencia | |
| Bioquímica | ||
| Creatinina | 0,47mg/dL | 0,5-0,9 |
| Resto de los parámetros bioquímicos dentro del rango de normalidad | ||
| Hemograma | ||
| Leucocitos | 18.080/μL (79,9% N) | 4-10 |
| Resto de parámetros dentro del rango de normalidad | ||
| Coagulación | ||
| Actividad de protrombina | 44% | 70-120 |
| INR | 1,85 | 0,88-130 |
| APTT | 31,7 s | 23-36 |
| Dímero D | 44.951μg/L | 45-500 |
| Resultados analíticos relevantes durante el ingreso | ||
|---|---|---|
| Resultado | Valores de referencia | |
| Homocisteína | 186μmol/L | 5-15 |
| Valores de ácido fólico y vitamina B12dentro de los rangos de normalidad | ||
| Anticuerpos anticardiolipinas | Negativo | |
| Anticuerpo lúpico | Negativo | |
| Estudio de la mutación G20210 del gen de la protrombina | Genotipo normal | |
| Estudio de la mutación C677 T del gen de la MTHFR | Genotipo normal | |
| Estudio de la mutación G1691A del gen del factor V | Heterocigota | |
| APCR | Positivo | |
| Aminoácidos libres en suero | ||
| Metionina | 653μmol/L | Niños: hasta 39 |
| Adultos: 11-43 | ||
| Resto de los aminoácidos dentro de los valores de referencia | ||
| Homocisteína en orina | 12μmol/g | Hasta 2 |
Se realizó estudio de trombofilia y se encontró déficit del factor VII (ya diagnosticado previamente), que era portadora de factor V de Leiden y de hiperhomocisteinemia severa: 186μmol/L (valores de referencia: 5-15μmol/L).
Se instauró tratamiento con vitaminas B1-B6-B12 y ácido fólico, que no se tradujo en un descenso de la homocisteína, por lo que, ante la sospecha de déficit de cistationina beta-sintetasa, se determinaron los aminoácidos en sangre y apareció hipermetioninemia, con valores de 653μmol/L (valores de referencia normales en niños hasta 39μmol/L). Se realizó estudio genético de las mutaciones más frecuentes en el gen CBS: I278T, G307S y T191M, con un resultado negativo para estas. Se solicitó entonces la secuenciación completa del gen y se identificó la presencia en homocigosis de la mutación patogénica c1058C>T en el gen CBS, una mutación missense que conduce a la sustitución de un aminoácido treonina por metionina en la posición 353 (T353M). Este resultado fue compatible con un fenotipo de homocistinuria clásica. Se realizó estudio familiar en el que apareció esta misma mutación en heterocigosis en ambos progenitores (que negaron consanguinidad) y en la hermana de la paciente.
Se instauró tratamiento dietético pobre en metionina y proteínas totales, suplementos vitamínicos, cistina y betaína, Xarelto y media de compresión. Evolucionó clínicamente sin signos de trombosis y mantuvo los valores de homocisteína por debajo de 80μmol/L.
DiscusiónSe conoce con el nombre de homocistinuria clásica a la debida a errores congénitos del metabolismo de la homocisteína. Presentamos un caso clínico de homocistinuria clásica, diagnosticada a partir de un episodio de trombosis venosa profunda. Tras varios estudios se encontró en homocigosis la mutación T353M en el gen CBS, asociada a homocistinuria clásica, con fenotipo no respondedor a B6.
En 1999 se describieron 92 mutaciones diferentes asociadas con la enfermedad en 310 alelos examinados14. El gen, localizado en el cromosoma 21q22.3, contiene 23 exones y una cuarta parte de las mutaciones se encuentran en el exón 3. Las 2más frecuentes, I278T y G307S, se encuentran en el exón 8 y T353M en el exón 1014.
En 2003, Kruger et al. estudiaron la relación entre los fenotipos bioquímicos y la clínica de 12 pacientes deficientes en cistationina beta-sintetasa, de 11 familias en Georgia (Estados Unidos). I278T y T353M representaron el 45% de las mutaciones encontradas. T353M, encontrada previamente en 4 pacientes afroamericanos, fue asociada con fenotipo no respondedor a B6. La actividad de T353M es <1,7% del wild type. Lo relacionaron con una forma severa de la enfermedad y proponen realizar un cribado directo de las mutaciones I278T y T353M en los pacientes homocistinúricos, por ser las más prevalentes, ya que un diagnóstico precoz mejoraría el pronóstico de la enfermedad15.
En España, la mutación T191M representó el 50% de los alelos mutantes. Urreizti et al. describen la prevalencia de T191M en pacientes diagnosticados de homocistinuria clásica, basándose en las manifestaciones clínicas sugestivas de la presencia en homocigosis de mutaciones en el gen CBS8. Los pacientes que presentaron la mutación T353M lo hicieron en heterocigosis compuesta T353M/T191M7,8, a diferencia de nuestro caso, en el que la paciente presentó la mutación T353M en homocigosis.
En julio de 2013, se aprobaron en el Pleno del Consejo Interterritorial del Sistema Nacional de Salud las enfermedades que forman parte del programa poblacional de cribado neonatal de enfermedades endocrino-metabólicas, incluido en la cartera común básica de servicios asistenciales del Sistema Nacional de Salud (Orden SSI/2065/2014, BOE 269, 6/11/2014). Estas enfermedades son hipotiroidismo congénito, fenilcetonuria, fibrosis quística, deficiencia de acil coenzima A deshidrogenasa de cadena media, deficiencia de 3-hidroxil acil-CoA deshidrogenasa de cadena larga, acidemia glutárica de tipo I y anemia falciforme. Además, incluyen el cribado neonatal de enfermedad de la orina con olor a jarabe de arce, la acidemia isovalérica y la homocistinuria dentro de un programa piloto.
El cribado neonatal de la homocistinuria, por tanto, permitiría la intervención precoz y un manejo adecuado de situaciones agudas, lo que mejoraría claramente el pronóstico de la enfermedad. Permitiría, a su vez, una pronta detección antes de su inicio, lo que evitaría la aparición de secuelas irreversibles y ofrecería la oportunidad de recibir consejo genético y la posibilidad de diagnóstico prenatal. En la actualidad varias comunidades autónomas (como Castilla-La Mancha, Murcia, Aragón, País Vasco, La Rioja, Andalucía, etc.) ya tienen implantando el cribado neonatal de homocistinuria.
ConclusiónLa prevalencia de la homocistinuria es baja, pero tiene una importante morbimortalidad asociada. En nuestro país, la mutación más frecuente es la T191M, sin embargo, existen otras como la T353M descritas en pacientes españoles. La paciente del caso clínico que exponemos presentó T353M en homocigosis, con una mayor morbimortalidad asociada, y creemos que habría que considerar esta mutación a la hora del diagnóstico de homocistinurias. La prevención de las complicaciones de esta enfermedad requiere su detección precoz para implantar un tratamiento que posibilite el mantenimiento de los niveles de homocisteína dentro de valores fisiológicos, mediante restricciones dietéticas y tratamiento farmacológico. Por ello, la homocistinuria se ha incluido en el cribado neonatal de metabolopatías existente en varias comunidades autónomas.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
