


El síndrome de Gitelman es una tubulopatía de herencia autosómica recesiva debida a mutaciones inactivantes en el gen SLC12A3 que codifica para el cotransportador sodio-cloro (NCC). El NCC es una proteína de membrana que pertenece a la familia de transportadores SLC12 cloro-catiónicos que constituye la principal vía de reabsorción de sodio y cloro (NaCl), determina la presión arterial y es el lugar de acción de los diuréticos tipo tiazida. El síndrome de Gitelman se caracteriza por hipopotasemia, hipomagnesemia, alcalosis metabólica, normocalcemia e hipocalciuria. El diagnóstico diferencial se realiza con el síndrome de Bartter tipoiii y la hipomagnesemia renal con hipocalciuria. Puede ser asintomático o expresarse con síntomas leves (calambres, fatiga o dolor articular) o con síntomas más graves (tetania, convulsiones). A pesar de considerarse benigno, la combinación de hipopotasemia con hipomagnesemia puede prolongar el intervalo QT y desencadenar arritmias que pueden amenazar la vida del paciente. Por todo ello resulta importante el diagnóstico diferencial y la confirmación mediante el estudio genético de cara al seguimiento de los pacientes y al asesoramiento genético.
Gitelman syndrome, an autosomal recessive tubulopathy, is caused by inactivating mutations in SLC12A3 gene. This gene codes for the sodium chloride co-transporter (NCC), a membrane protein that belongs to the family of SLC12 chloride-cationic transporters. NCC constitutes the main route of sodium chloride (NaCl) reabsorption, determines blood pressure, and is the site of action of thiazide-type diuretics. Gitelman syndrome usually involves hypokalaemia, hypomagnesaemia, metabolic alkalosis, and hypocalciuria. The differential diagnosis for Gitelman syndrome includes Bartter syndrome typeiii and renal hypomagnesaemia. Symptoms reported in the literature range from asymptomatic, to mild symptoms of cramps and fatigue, to severe manifestations such as tetany and seizures. The prognosis is generally good, but a few patients with hypokalaemia and hypomagnesaemia may have a prolonged QT interval and trigger potentially life-threatening arrhythmias. Thus, genetic testing is important to confirm the diagnosis, as well as in the follow-up of patients.
Las enfermedades del túbulo renal, o tubulopatías, se definen como alteraciones clínicas en las que existe una disfunción tubular específica con afectación escasa o nula de la función glomerular en estadios precoces. Se clasifican en tubulopatías primarias o hereditarias y tubulopatías secundarias debidas a fármacos, tóxicos u otras enfermedades1,2. El síndrome de Gitelman, también llamado hipokalemia-hipomagnesemia familiar, es una tubulopatía primaria que se transmite mediante herencia autosómica recesiva. Se debe a mutaciones en el gen solute carrier family 12, member 3 (SLC12A3) (OMIM #263800), cromosoma 16q13, que codifica el cotransportador del túbulo distal Na/Cl sensible a las tiazidas, también conocido como thiazide sensitive Na-Cl cotransporter (TSC) o sodium-chloride co-transporter (NCCT)1-3. Recientemente se han identificado mutaciones en el gen CLCNKB (OMIM #602023), cromosoma 1p36, que codifica para el canal de cloro (Chloride Channel, Voltage-Sensitive Kb [CIC-Kb]) como causa de síndrome de Gitelman en una minoría de pacientes4. Este síndrome se caracteriza por alcalosis metabólica, presión arterial normal con hipopotasemia, hipomagnesemia e hipocalciuria. La presentación clínica es muy heterogénea, así como la edad de inicio, desde pacientes pediátricos con manifestaciones muy claras hasta adultos con síntomas muy leves o asintomáticos. Su prevalencia estimada es de 1/1.000.000 de habitantes. El diagnóstico diferencial se realiza con el síndrome de Bartter tipoiii y la hipomagnesemia renal, con hipocalciuria. La confirmación se realiza mediante el estudio genético2,4,5. Presentamos un caso de diagnóstico casual en un paciente pediátrico que acude a urgencias por vómitos.
Caso clínicoNiña de 4 años de edad que acude al servicio de urgencias del hospital por presentar un cuadro de vómitos de más de 24h. Los padres no refieren la presencia de fiebre, pero sí cefaleas y dolor abdominal epigástrico. Sin antecedentes personales de interés, en los antecedentes familiares destaca una tía materna con síndrome de Gitelman.
En la exploración física presenta un estado general conservado, sin hallazgos significativos. Los resultados más destacados de la analítica solicitada en el servicio de urgencias se muestran en la tabla 1. Los hallazgos más significativos en dicha analítica son la presencia de una ligera alcalosis en la gasometría venosa (pH7,48), hipopotasemia severa (1,98mEq/l) y presencia en la tira de orina de cuerpos cetónicos++++ y proteínas++. Se decide el ingreso de la paciente para su estudio y tratamiento.
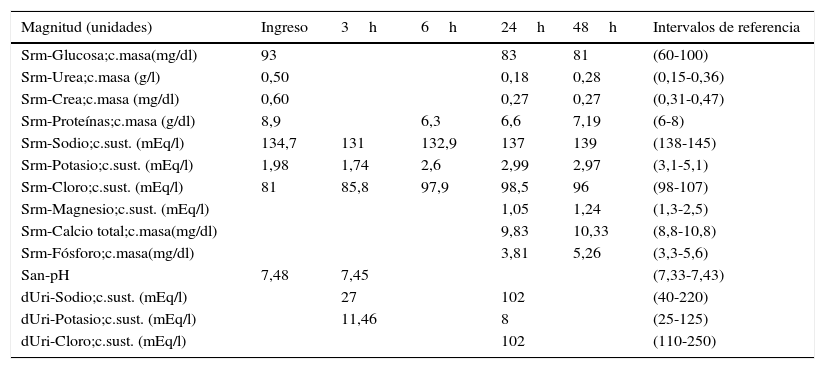
Resultados de las magnitudes analíticas urgentes y programadas de la paciente
| Magnitud (unidades) | Ingreso | 3h | 6h | 24h | 48h | Intervalos de referencia |
|---|---|---|---|---|---|---|
| Srm-Glucosa;c.masa(mg/dl) | 93 | 83 | 81 | (60-100) | ||
| Srm-Urea;c.masa (g/l) | 0,50 | 0,18 | 0,28 | (0,15-0,36) | ||
| Srm-Crea;c.masa (mg/dl) | 0,60 | 0,27 | 0,27 | (0,31-0,47) | ||
| Srm-Proteínas;c.masa (g/dl) | 8,9 | 6,3 | 6,6 | 7,19 | (6-8) | |
| Srm-Sodio;c.sust. (mEq/l) | 134,7 | 131 | 132,9 | 137 | 139 | (138-145) |
| Srm-Potasio;c.sust. (mEq/l) | 1,98 | 1,74 | 2,6 | 2,99 | 2,97 | (3,1-5,1) |
| Srm-Cloro;c.sust. (mEq/l) | 81 | 85,8 | 97,9 | 98,5 | 96 | (98-107) |
| Srm-Magnesio;c.sust. (mEq/l) | 1,05 | 1,24 | (1,3-2,5) | |||
| Srm-Calcio total;c.masa(mg/dl) | 9,83 | 10,33 | (8,8-10,8) | |||
| Srm-Fósforo;c.masa(mg/dl) | 3,81 | 5,26 | (3,3-5,6) | |||
| San-pH | 7,48 | 7,45 | (7,33-7,43) | |||
| dUri-Sodio;c.sust. (mEq/l) | 27 | 102 | (40-220) | |||
| dUri-Potasio;c.sust. (mEq/l) | 11,46 | 8 | (25-125) | |||
| dUri-Cloro;c.sust. (mEq/l) | 102 | (110-250) |
Los resultados de la bioquímica en suero y orina se muestran en la tabla 1. La hipopotasemia se define como la existencia de una concentración de potasio (K) plasmático inferior a 3,5mEq/l, leve de 3-3,5mEq/l, moderada de 2,5-3mEq/l y severa cuando es menor de 2,5mEq/l. La paciente presenta vómitos de larga evolución y se inicia un tratamiento con perfusión intravenosa de electrólitos, normalizándose el ionograma, a excepción del K. Ante una hipopotasemia confirmada se debe realizar una historia clínica detallada, valorar el volumen extracelular (VEC), los iones (Na, K y Cl) en suero y en orina, incluyendo la determinación de magnesio (Mg) y la gasometría junto con un estudio hormonal (aldosterona y renina). Entre las múltiples causas de una hipopotasemia, algunas tienen su origen en un defecto renal primario que impide la retención de potasio por parte de este órgano, por lo que se debe realizar también el estudio de la función renal, glomerular y tubular6,7.
En este caso se descarta la toma subrepticia de diuréticos. Se debe sospechar el síndrome de Bartter y el síndrome de Gitelman, ambos tubulopatías hereditarias, cuando se presenta una hipopotasemia con alcalosis metabólica y los electrólitos urinarios Na, K y Cl son elevados o inadecuados para el VEC, que puede estar bajo o normal.
Estos síndromes presentan pérdida renal de sales, alcalosis metabólica hipopotasémica, hiperaldosteronismo hiperreninémico, presión arterial normal e hiperplasia del aparato yuxtaglomerular8-10.
El diagnóstico diferencial entre el síndrome de Gitelman y el síndrome de Bartter (tipoiii) se basa en la excreción urinaria de calcio: en el síndrome de Gitelman está disminuida, mientras que en el síndrome de Bartter es normal o está aumentada. Respecto al diagnóstico diferencial con la hipomagnesemia renal con hipocalciuria, se diferencian en que la concentración de potasio se mantiene en valores dentro del rango de referencia.
Los criterios diagnósticos del síndrome de Gitelman11 en pacientes con presión arterial normal y en ausencia de ingesta de diuréticos son:
- a)
Hipomagnesemia de origen renal (Mg<1,6mg/dl, en presencia de magnesuria inapropiadamente elevada, excreción fraccional [EF] de Mg>9%).
- b)
Hipopotasemia de origen renal (K<3,6mEq/l, en presencia de una concentración de potasio urinario inapropiadamente elevado, EF K>16%).
- c)
Excreción urinaria de calcio <2mg/kg/24h (raramente superior a 0,5mg/kg/24h).
Los análisis más significativos, bioquímica en suero, en orina, función glomerular y tubular se muestran en las tablas 1 y 2. Se confirma la hipopotasemia, se observa hipomagnesemia e hipocalciuria. Los resultados de la función glomerular están dentro de los rangos de referencia.
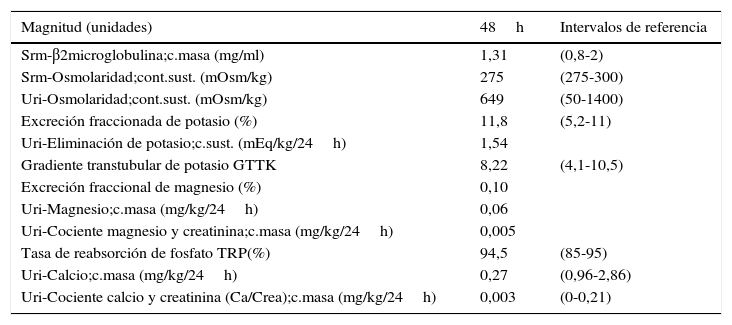
Magnitudes de función renal
| Magnitud (unidades) | 48h | Intervalos de referencia |
|---|---|---|
| Srm-β2microglobulina;c.masa (mg/ml) | 1,31 | (0,8-2) |
| Srm-Osmolaridad;cont.sust. (mOsm/kg) | 275 | (275-300) |
| Uri-Osmolaridad;cont.sust. (mOsm/kg) | 649 | (50-1400) |
| Excreción fraccionada de potasio (%) | 11,8 | (5,2-11) |
| Uri-Eliminación de potasio;c.sust. (mEq/kg/24h) | 1,54 | |
| Gradiente transtubular de potasio GTTK | 8,22 | (4,1-10,5) |
| Excreción fraccional de magnesio (%) | 0,10 | |
| Uri-Magnesio;c.masa (mg/kg/24h) | 0,06 | |
| Uri-Cociente magnesio y creatinina;c.masa (mg/kg/24h) | 0,005 | |
| Tasa de reabsorción de fosfato TRP(%) | 94,5 | (85-95) |
| Uri-Calcio;c.masa (mg/kg/24h) | 0,27 | (0,96-2,86) |
| Uri-Cociente calcio y creatinina (Ca/Crea);c.masa (mg/kg/24h) | 0,003 | (0-0,21) |
La β2-microglobulina, marcador de la función tubular, obtuvo valores dentro de los rangos de referencia. El estudio hormonal muestra valores de renina y angiotensina por encima de los rangos de referencia, ya que se produce hiperaldosteronismo secundario a la pérdida de NaCl.
El valor del gradiente transtubular de K, GTTK o TTKG (Transtubular K Gradient) es superior a 7 (8,22), indicando la presencia de actividad mineralocorticoide en el túbulo contorneado distal6,7. La excreción fraccionada de potasio (EFK) es del 11,8%, elevada, aunque no llega a ser >16%. Presenta hipomagnesemia con un Mg en suero de 1,51mg/dl (<1,6mg/dl) con una magnesuria de <9%. La calcemia es normal y la calciuria es de 0,27mg/kg/24h (<0,5mg/kg/24h), por lo que está muy disminuida.
La proteína C reactiva y el estudio de inmunidad (IgG, IgA, IgM, IgE) obtuvieron valores dentro del rango de referencia. Los anticuerpos para el estudio de la celiaquía y los análisis microbiológicos (coprocultivo, virus en heces y urocultivo) resultaron negativos. Se descarta gastroenteritis aguda por la limitación del cuadro y los análisis microbiológicos negativos.
Pruebas de imagen y pruebas funcionales: radiografía de carpo acorde con la edad cronológica, no se aprecian signos de raquitismo; ecografía renal y suprarrenal: riñones de tamaño y morfología normal con buen índice corticomedular, vejiga sin hallazgos; ecocardiograma: normal; ECG: rítmico, sinusal, alargamiento del QT (0,45).
La confirmación del síndrome de Gitelman se realiza siempre con el estudio genético, que consiste en una amplificación y secuenciación completa del gen SLC1A3. El resultado del estudio genético confirma el diagnóstico de síndrome de Gitelman: la paciente es homocigota para la mutación intrón 9+1G>T; esta mutación específica se ha descrito en la etnia gitana a la que pertenece la paciente3.
El diagnóstico definitivo es síndrome de Gitelman con hipopotasemia y aplanamiento de la onda T. Se establece el tratamiento con perfusión intravenosa con electrólitos y posteriormente con suplementos vía oral, normalizándose el ionograma pero manteniendo la hipopotasemia. Durante el ingreso permanece afebril con constantes dentro de la normalidad, buen estado general y buena tolerancia oral. Se le da el alta días después con las recomendaciones terapéuticas: dieta rica en potasio y magnesio; medicación, suplementos de potasio y magnesio.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
