


El cobre es un elemento traza esencial (oligoelemento) necesario para la adecuada acción de diversas enzimas, ferroxidasa (EC 1.16.3.1), también denominada ferroxidasa 1 o, más comúnmente, ceruloplasmina, citocromo-c oxidasa (EC 1.9.3.1), superoxidodismutasa (EC 1.15.1.1.), dopamina-b-hidroxilasa (EC 1.14.17.1), etc.1,2. El cobre es un elemento traza esencial que actúa como cofactor de transferencia electrónica de diversas cuproenzimas; participa en procesos vitales como la oxidación del hierro, la respiración celular, la eliminación de radicales libres, la biosíntesis de catecolaminas y melanina, así como en la formación del tejido conectivo. Además, aunque es un micronutriente esencial, en situaciones de exceso genera radicales libres altamente tóxicos para el organismo.
El cobre se absorbe principalmente en el estómago y el duodeno, y es transportado por la vena porta (unido a la albúmina, la transcupreína y los aminoácidos, principalmente la histidina) hacia el hígado, su principal órgano homeostático, donde una parte de la cantidad que llega es incorporada a la ferroxidasa y otra parte es excretada por la bilis3. Muchos parámetros afectan a la absorción del cobre, incluyendo la edad, el género y el tipo de alimentación, oscilando esta entre un 12 y un 71%4.
Alrededor de un 90% del cobre circulante en la sangre se encuentra unido a la ceruloplasmina y aproximadamente un 10% unido a proteínas transportadoras (albúmina o transcupreína)5. Otros artículos2 coinciden en estos porcentajes. Este hecho es relevante dado que la mayoría de los métodos de análisis miden cobre total, por lo que los aumentos o descensos de la ceruloplasmina son cruciales a la hora de interpretar los resultados.
La excreción de cobre se hace por la bilis en un 99%, siendo la cantidad excretada directamente proporcional a la reserva hepática del metal. En la bilis, el cobre está como un complejo no absorbible. No presenta circulación enterohepática, por lo que prácticamente el cobre eliminado equivale al absorbido por la dieta.
Los grupos de alimentos tales como frutos secos, vísceras (hígado…), crustáceos (marisco) y, en menor extensión, cereales y frutas pueden ser considerados una apropiada fuente de cobre, mientras que la leche y los lácteos contiene una mínima proporción. La cantidad mínima requerida en la dieta para alcanzar un adecuado balance oscila entre 800 y 2.400μg/día. La EFSA, Autoridad Europea de Seguridad Alimentaria, recomienda una ingesta diaria adecuada de 1.600μg/día (0,024mmol/día)6.
Las concentraciones séricas elevadas de cobre en el suero se asocian a intoxicaciones1,2. El cobre, dado que es un reactante de fase aguda, se eleva en inflamación, infección, tumores malignos, el embarazo, y otros factores de estrés biológicos5. La deficiencia de cobre puede dar lugar a niños malnutridos, bebés prematuros, enfermedad cardiovascular, síndromes de malabsorción, anemia microcítica, neutropenia, anomalías de la pigmentación, debilidad muscular. Su déficit puede producir anemia hipocrómica y microcítica, alteraciones neurológicas, defectos de la queratinización y de la formación de cartílago y osificación, anomalías de la pigmentación de la piel, y otras1. Las causas más frecuentes de déficit de cobre son los síndromes de malabsorción intestinal, estados de malnutrición en general, el síndrome nefrótico, donde se pierden grandes cantidades de proteínas, entre ellas, la ferroxidasa (ceruloplasmina), la cirugía bariátrica o bypass gástrico7,8y el exceso de cinc9–11.
El laboratorio clínico ejerce un papel esencial en el diagnóstico y la monitorización de síndromes y enfermedades fundamentadas en el déficit o exceso de cobre en el organismo, entre las que destacan: la enfermedad de Wilson (incapacidad para secretar cobre unido a ferroxidasa desde el hígado)12–17, la cirrosis biliar primaria, la enfermedad de Menkes (defecto genético del transporte y almacenamiento de cobre ligado al cromosoma X)5,18 y el síndrome del cuerno occipital (su forma leve, más conocida, es también llamada síndrome de Ehlers-Danlos tipo 9)19,20. De todas las anteriores, hay que resaltar el papel del laboratorio en el diagnóstico de la enfermedad de Wilson, trastorno hereditario del metabolismo del cobre, caracterizado por un defecto de su excreción biliar que conduce a su acumulación en el organismo, principalmente en el hígado y el encéfalo, con efectos tóxicos por daño oxidante. Actualmente, esta enfermedad sigue siendo un desafío diagnóstico debido a que es una patología poco común, en la que la forma y la edad de presentación pueden ser heterogéneas, con manifestaciones clínicas inespecíficas y para la que existen diversas pruebas diagnósticas en el laboratorio. Cuando un clínico se halla a un paciente con enfermedad hepática y/o neurológica no explicable por otras causas, no debe olvidarse de valorar un posible caso de enfermedad de Wilson. Por ello es importante realizar un diagnóstico correcto y temprano, donde el laboratorio clínico, a fin de poder ayudar en el proceso del diagnóstico, debe establecer unos procedimientos de medida de cobre sérico y urinario adecuados.
Para evaluar el estado funcional de cobre del organismo se han propuesto diversidad de métodos indirectos (entre ellos la medida de la actividad catalítica y/o masa de enzimas como la ferroxidasa, actividad de la superóxido-dismutasa, la citocromo-c oxidasa, lisil oxidasa…). Por otro lado, los métodos directos que miden la concentración o el contenido de cobre en diversos especímenes (suero, orina, tejido)21 son los que se consideran en esta revisión.
Objeto y campo de aplicaciónDescribir los métodos analíticos para la medida directa del contenido de cobre en diversos especímenes biológicos y recomendar los procedimientos que presentan una mayor fiabilidad dentro de los que son más accesibles al laboratorio clínico.
Revisión metodológicaEspectrometría de absorción molecularLa espectrometría de absorción molecular fue uno de los primeros métodos analíticos, desarrollados durante los años 50 y 60, para medir la concentración de cobre en el suero. Posteriormente, fue adaptado para realizar la determinación de cobre en orina y otros tejidos. Se basa en la absorción del complejo formado por el cobre (i+ii) con un agente quelante.
Entre estos, los más empleados fueron el dietilditiocarbamato, la 4,7-difenil-1,10-fenantrolina, el 2,9-dimetil-4,7-difenil-1,10-fenantrolina (batocuproína), la bisciclohexanonaoxalil dihidrazona (cuprizona) y la oxalildihidrazida. Los 2 últimos presentan la mayor sensibilidad analítica (mayor absortividad molar) pero hay que cuidar más las condiciones analíticas debido a la mayor facilidad de contaminación. El método de la batocuproína es el más específico de todos estos, manteniendo una sensibilidad analítica aceptable dentro de la metodología de absorción molecular.
Voltametría de disolución anódicaEste método se basa en la deposición, por reducción, de los iones de cobre en forma de cobre metálico sobre el ánodo (electrodo de mercurio). Tras ello el potencial se hace más positivo y se redisuelve este cobre (reoxidación) pasando de nuevo a la solución, lo cual produce una corriente proporcional a la concentración de cobre en la muestra. Este método se ha aplicado a la medida de la concentración de cobre en el suero pero está poco extendido y no ha sido validado suficientemente, excepto en análisis ambientales. Tiene la ventaja de poder medir la concentración de varios elementos al mismo tiempo debido a que el potencial del par ion/metal es característico de cada metal.
Activación neutrónicaSe fundamenta en la transformación del 63Cu en 64Cu inestable por bombardeo con neutrones. El 64Cu tiene una semivida de 12,9 h y se desintegra a 64Ni estable emitiendo radiación gamma de 1,34 MeV. Tras la irradiación hay que separar químicamente el cobre ya que interfiere la radiación de 1,37 MeV del 23Na. Permite el análisis de cobre en suero, orina y tejidos, pero es laborioso y requiere un equipo costoso con personal muy cualificado. Se consideraba que era el método definitivo para la medida de la concentración o contenido de cobre.
Espectrometría de absorción atómica de llamaEl campo de la espectrometría de absorción atómica (AAS) ha visto en la década pasada la comercialización de la AAS de alta resolución con fuente continua. Esta tecnología proporciona una serie de ventajas sobre la convencional AAS con fuente de línea. Una ventaja muy evidente es que ya no es necesario, con estos instrumentos, la utilización de las lámparas de cátodo hueco (o de descarga sin electrodos), dado que con una sola lámpara (de arco corto de xenón) pueden medirse la mayoría de los elementos, incluso los no metálicos22. Actualmente, en la mayoría de los laboratorios clínicos se dispone de la tecnología convencional con fuente de línea y por ello es esta la tecnología que se detalla en el presente documento.
Se basa en que la energía de una llama de aire/acetileno hace que los átomos de cobre de una disolución se encuentren en estado fundamental y sean entonces capaces de absorber la radiación de 324,7nm proveniente de una lámpara de cátodo hueco de cobre. Mediante esta técnica puede medirse la concentración o el contenido de cobre en el suero, orina y tejidos. Por sus características, es la técnica de elección para medir la concentración de cobre en el suero en ausencia de un método de referencia. La línea de resonancia del cobre está lejos de las de otros elementos que habitualmente se encuentran en el suero. El límite de detección se sitúa alrededor de 0,6μmol/L (38,12μg/L), que es más que suficiente para la medición de las concentraciones en suero (concentraciones con valores alrededor de 11,0 a 25,0μmol/L [698,9 a 1.588,5μg/L]), con lo que fácilmente se pone de manifiesto el déficit de este elemento. Se han descrito múltiples métodos de preparación previa de la muestra desde los años 70, pero actualmente se tiende a la menor manipulación posible de la misma, evitando pasos previos de desproteinización o de extracción con disolventes orgánicos.
La medida de la concentración de cobre en la orina puede realizarse por AAS de llama (FAAS) pero el método de elección es la AAS con atomización electrotérmica y corrección de fondo por efecto Zeeman, ya que la primera no permite diferenciar entre concentraciones fisiológicas y bajas de este elemento23. Los métodos que se han descrito para el análisis de orina por llama miden la concentración de cobre en orina bien directamente, tras concentración o tras extracción de la misma. Los 2 primeros métodos tienen el problema del alto contenido en sales de las muestras que puede afectar a los resultados. La extracción puede ser fuente de interferencias o contaminaciones. En caso de analizar las muestras de orina por esta técnica, es aconsejable preparar los patrones en una matriz salina similar a las de las orinas.
La medida del contenido de cobre en tejido, en especial hepático, por técnica de llama presenta el inconveniente de no diferenciar entre valores normales y bajos. En general, esto puede no ser un gran inconveniente ya que habitualmente se mide el contenido de cobre en el hígado para el diagnóstico de la enfermedad de Wilson en donde los contenidos son, al menos, 5 veces superiores al intervalo de referencia.
El contenido de cobre en el pelo puede medirse mediante esta técnica, pero es un espécimen que genera gran controversia dado que dicho contenido de cobre puede ser muy variable, 10-100 veces más elevado que en suero e influido por factores como el sexo, el uso de anticonceptivos, situaciones patológicas asociadas a redistribución del cobre entre plasma y tejidos. Además, puede encontrarse alterado por la utilización de cosméticos, productos de higiene y contaminación ambiental, entre otros1. Esto hace necesario un cuidadoso lavado del cabello previo a la toma de la muestra teniendo especial cuidado de no arrastrar algo del cobre capilar. Por ello, esta matriz, aunque presenta la ventaja de su fácil recolección y conservación, requiere de una adecuada técnica de lavado y preparación de la muestra a fin de evitar contaminaciones externas.
Espectrometría de absorción atómica con atomización electrotérmicaEn este apartado, al igual que en el anterior, se hace referencia al método convencional con fuente de línea. Con la técnica de espectrometría de absorción atómica con atomización electrotérmica (ETAAS) se consiguen límites de detección más bajos que con la de llama (< 0,6μmol/L) debido a una serie de factores entre los que se encuentran un mayor control de la temperatura de atomización, un mayor tiempo de residencia de la nube atómica en el paso óptico, una menor influencia del fondo, etc.22,23.
Debido a la concentración de este elemento en el suero, debe hacerse una dilución previa del mismo de, al menos, 1:15, lo que puede aumentar la imprecisión de la medida, por lo que, como se indicó en el apartado anterior, para su medida se prefiere la técnica de llama, más rápida que la de atomización electrotérmica.
La técnica más adecuada para una medición fiable de la concentración de cobre en orina es la de atomización electrotérmica con corrección de fondo por efecto Zeeman longitudinal (el empleo de una corrección de fondo por Zeeman transversal produce pérdidas de sensibilidad analítica de alrededor del 50%, pero aún así la calidad de corrección de fondo del efecto Zeeman frente a la lámpara de deuterio es mejor)23.
La medida del contenido de cobre en un tejido por absorción atómica con atomización electrotérmica permite detectar con fiabilidad los valores fisiológicos.
En el diagnóstico de la enfermedad de Wilson es interesante realizar la medida de la concentración de cobre sérico no unido a ferroxidasa por ETAAS, junto con la concentración catalítica de la ferroxidasa en suero. El método se basa en la extracción del cobre no unido con metil-isobutil-cetona tras su acomplejamiento con amino-pirrolidin-ditiocarbamato24. Este método es laborioso. Puede emplearse una estimación indirecta:
[Cobre unido a ferroxidasa (μmol/L)]=[ferroxidasa (μmol/L)]×0,05
[Cobre no unido a ferroxidasa]=[cobre] – [cobre unido a ferroxidasa]
Los métodos calculados (ecuaciones) que pueden consultarse en la literatura científica aportan unos valores discriminantes en los que habría que considerar cuál es el método para la medida tanto de la ceruloplasmina como del cobre y de la variabilidad analítica de ambos, lo que puede hacer que la ecuación mencionada anteriormente sea difícilmente extrapolable para laboratorios clínicos que utilizan métodos analíticos distintos. Además, estas ecuaciones proporcionan un alto porcentaje de resultados negativos, lo que ciertamente incrementa las reticencias en su utilización25,26. A fin de subsanar las evidentes limitaciones de las estimaciones indirectas anteriores, se han desarrollado distintas aproximaciones analíticas que midan directamente la fracción de cobre no unida a ceruloplasmina, como la determinación del cobre ultrafiltrable (cobre libre)27, que requiere la ultrafiltración del plasma a través de una membrana con la capacidad de retener proteínas como la albúmina, la ceruloplasmina y la transcupreína. La complejidad analítica de estos y los muy exigentes requerimientos preanalíticos hacen difícil su implementación clínica. Máxime cuando parece que únicamente podría ser potencialmente de utilidad para monitorizar a pacientes con enfermedad de Wilson en tratamiento con cinc. En los últimos años, Squitti28 afirma haber patentado un método fluorescente basado en la cumarina, capaz de medir únicamente el cobre no unido a ceruloplasmina, método que puede adaptarse a autoanalizadores convencionales, aunque incorpora un paso previo de extracción en fase sólida que complica su aplicabilidad práctica.
Espectrometría de masas con plasma de acoplamiento inducidoLa espectrometría de masas con plasma de acoplamiento inducido (ICP-MS) es una técnica de reciente incorporación al laboratorio clínico. Quizás debido a su complejidad y coste elevado no está implementada de forma mayoritaria. Mediante esta técnica es posible determinar de forma cuantitativa la mayoría de los elementos de la tabla periódica a niveles de traza y ultratraza, partiendo de muestras en disolución acuosa. La ICP-MS utiliza como fuente de radiación un plasma de acoplamiento inducido y como detector un espectrómetro de masas29. Los iones generados en el plasma pasan a través de un filtro cuadrupolar, que separa los iones generados en el plasma según su relación m/z. Cada una de las masas separadas llega al detector, donde se cuantifica el elemento de la muestra. Para controlar la estabilidad de la señal obtenida y para corregir interferencias no espectrales se emplean los patrones internos. Estos son elementos que no están presentes en la muestra: indio, rodio, itrio, escandio, germanio, bismuto, etc.; se añaden tanto al blanco como a los estándares y a las muestras.
Existen varios tipos de interferencias en ICP-MS:
- –
Espectroscópicas: surgen cuando una especie iónica tiene una relación m/z igual a la del analito. Se evitan con una adecuada selección del isótopo a analizar.
- –
No espectroscópicas: debidas a la matriz, que suelen provocar una disminución de la señal al repelerse iones en el haz procedente del plasma. Pueden corregirse diluyendo la muestra, separando previamente los compuestos interferentes, introduciendo la muestra de forma diferente o bien empleando un patrón interno.
Pese a la limitación del encarecimiento que supone el ICP-MS para un laboratorio clínico, presenta una serie de ventajas que se deben tener en cuenta: mejores límites de detección (< 1μg/L o 0,02μmol/L) que la FAAS y la ETAAS. Se obtienen espectros sencillos y fácilmente interpretables y permite realizar un análisis simultáneo multielemental de casi todos los elementos del sistema periódico. Es una técnica sensible y precisa, si bien las muestras que se introducen en un ICP-MS deben ser muy «limpias», evitando así que se obstruya el sistema de aspiración, la interfase o que aparezcan depósitos en el sistema óptico29.
Otras técnicasLa polarografía diferencial, la espectrofluorimetría de rayos X30, la espectrometría de emisión por arco y la espectrometría de emisión de plasma acoplado a radiofrecuencias no se han considerado aquí debido a que son técnicas muy específicas, de baja o nula implantación en el laboratorio clínico, precisan equipos caros y complejos y proporcionan resultados similares a los encontrados con las técnicas descritas.
Procedimientos analíticos recomendadosLa metodología más apropiada para la medida de cobre en especímenes biológicos es la AAS. En el caso de especímenes séricos es adecuada la técnica de llama, FAAS, mientras que para la cuantificación en orina e hígado es necesario utilizar la ETAAS. La FAAS y la ETAAS son las más difundidas actualmente en los laboratorios clínicos, ya que presentan sensibilidad suficiente y los equipos son menos costosos y, además, son de fácil manejo21. La FAAS es una técnica muy reproducible y rápida, aunque su sensibilidad es menor. La ETAAS es una técnica muy sensible, con un límite de detección, «limit of detection» (LOD) del orden de μg/l, pero es una técnica más lenta y que presenta interferencias. Principalmente se emplea en la detección del cobre en orina e hígado.
Las técnicas de espectrometría de absorción molecular deberían ser abandonadas en beneficio de estas últimas. Por otro lado, el ICP-MS es una técnica multielemental, muy sensible, con un LOD inferior a 1μg/L, del orden de ng/L. Es la técnica de elección cuando se requiere un análisis de múltiples elementos. Generalmente, en estos casos, el cobre se determina conjuntamente con el cinc y el selenio21,29.
Como ya se ha comentado anteriormente, presenta el inconveniente del coste elevado, manejo por personal con experiencia y de la presencia de ciertas interferencias. Pese a sus limitaciones, mantiene una serie de ventajas que han facilitado su introducción en los laboratorios en los últimos años. Es una técnica sensible y precisa que requiere un mínimo pretratamiento de la muestra y permite un análisis simultáneo multielemental. Asimismo, presenta un amplio rango dinámico y mejores límites de detección (< 1μg/L o 0,02μmol/L) que la ETAAS y la FAAS, obteniéndose con ella espectros sencillos y fácilmente interpretables31.
Obtención de especímenes en suero y orinaBiomarcadores del estado nutricional del cobreEl biomarcador adecuado debería tener la habilidad de detectar variaciones muy rápidas en los niveles de cobre antes de que aparecieran manifestaciones clínicas, así como la capacidad de predecir este hecho con una elevada sensibilidad y especificidad. El estado nutricional del cobre se ha valorado en las últimas décadas mediante la determinación del elemento en suero, plasma, orina y pelo. Las medidas de cobre en suero son las más utilizadas y, pese a que no se ha reconocido como el excelente biomarcador del estado nutricional de este elemento, el cobre en suero se considera buen indicador del estatus de cobre y de su ingesta. Así, Harvey et al., tras una exhaustiva revisión metodológica, indican la utilidad del cobre en suero, tanto en casos de depleción de cobre como en casos de exceso, teniendo en cuenta la influencia de factores como la edad, el género t el embarazo32. No ocurre lo mismo con el cobre en plasma, que, además de tener valores inferiores a los de suero, no parece capaz de evidenciar el exceso de cobre1.
Procedimientos preanalíticosLa contaminación es el principal problema en la medición de elementos traza. Hay que tener especial cuidado en la obtención y la manipulación de las muestras, puesto que estos elementos están presentes en el organismo en bajas concentraciones. En la guía National Committee for Clinical Laboratory (NCCLS) se detallan los procedimientos que deben tenerse en cuenta en la fase preanalítica33.
La Sociedad Española de Bioquímica Clínica y Patología Molecular (SEQC) también ha publicado recientemente los requerimientos técnicos y ambientales34 y las medidas de seguridad35 que se deben tener en cuenta en los laboratorios clínicos para la determinación de elementos traza. Además de los requerimientos recomendados para cada uno de los especímenes en particular, será necesaria la utilización de patrones y reactivos de alto grado de pureza.
Asimismo, el agua empleada debe presentar una calidad adecuada que asegure la fiabilidad de los resultados. Por ello, debería utilizarse agua ultrapura, tipo i (según los estándares de la American Society for Testing of Materials); conductividad 0,056μS/cm y resistividad 18MΩ×cm.
SueroSe recomienda el empleo de suero obtenido en tubos de vacío específicos para elementos traza (una vez comprobado que no presentan concentraciones detectables de cobre) o bien con jeringa de plástico. Deben evitarse los tubos de vacío con gel separador. Se permite un cierto grado de hemólisis, ya que el cobre se reparte en partes aproximadamente iguales entre el suero y los eritrocitos. Es recomendable realizar las extracciones a la misma hora del día (p. ej., entre las 08:00 y 10:00 horas) debido al ritmo circadiano que presentan las concentraciones de este elemento en el suero. Una vez extraída la sangre y coagulada, se centrifuga durante 10 min a 1.000-1.200g con el tapón puesto en el tubo para evitar contaminaciones procedentes de la centrífuga y las evaporaciones33. El suero puede conservarse en tubos de polipropileno o poliestireno bien tapados durante al menos 15 días refrigerados, o de manera prácticamente indefinida a –20°C. Todo el material que ha de estar en contacto con el espécimen (tubos de ensayo, puntas de pipeta, contenedores, etc.) ha de lavarse previamente, dejarlo entre 12 y 24 h en una solución de ácido nítrico 1 o 2mmol/L y enjuagarlos varias veces con agua destilada y desionizada antes de su uso.
OrinaLas orinas deben de recogerse idealmente durante 24 h, ya que las concentraciones de cobre en muestras de orina aisladas presentan una elevada variabilidad intraindividual. Es muy importante obtener una adecuada diuresis para la correcta valoración de la concentración de cobre en orina de 24 h. Es recomendable que el material empleado sea lavado con ácido previamente. A fin de mantener la estabilidad de las muestras, se podrían acidificar las muestras al recepcionarlas en el laboratorio. Asimismo, es esencial evitar los errores en la recogida de orina de 24 h, así como la contaminación del contenedor de muestra con cobre exógeno.
Una vez mezcladas las mismas, se alícuotan y conservan refrigeradas o congeladas a –20°C hasta su procesamiento. Si se han sometido a congelación, tras descongelarlas es necesario homogeneizarlas bien y centrifugarlas.
La excreción de cobre en orina refleja la concentración de cobre sérico no unido a ceruloplasmina en la circulación. La determinación de cobre en orina de 24 h puede ser útil tanto en el diagnóstico como en la monitorización del tratamiento (p. ej., en la enfermedad de Wilson).
Procedimiento de medida de cobre en suero por espectrometría de absorción molecular utilizando batocuproínaCondiciones analíticasMedir la absorbancia de muestras y patrón a 480nm frente a un blanco con ácido clorhídrico (HCl), ácido tricloroacético y cromógeno en las mismas proporciones que la muestra y agua en lugar del sobrenadante de la desproteinización.
Preparación de muestras y patronesTomar 2mL de suero y mezclarlos con 1mL de HCl 1mol/L. Dejar 20 min a temperatura ambiente y añadir 1mL de ácido tricloroacético 1,23mol/L; mezclar y centrifugar. Mezclar 2mL de sobrenadante con la solución cromogénica (disulfonato de batocuproína 0,53mmol/L, acetato sódico 3,3mol/L, pirosulfito de sodio 0,1mol/L, p-metilaminofenol 5,5mmol/L). Como patrón se emplea una solución acuosa de 31,5μmol/L de cobre.
Características metrológicasEl procedimiento es lineal hasta 126μmol/L y su detectabilidad en el suero es del orden de 3μmol/L. La imprecisión intraserial es inferior al 10% y la interserial al 15%. Es un procedimiento de fácil acceso por parte de cualquier laboratorio pero tiene la desventaja de que interfiere la hemólisis, precisa cantidades de suero elevadas (1-2mL), se requiere desproteinización previa del suero y digestión de la orina, y su análisis consume mucho tiempo. Este procedimiento puede automatizarse tras la desproteinización.
Procedimiento de medida de la concentración de cobre en suero por espectrometría de absorción atómica de llamaCondiciones analíticasSe emplea un espectrómetro de absorción atómica de llama. La lámpara de cátodo hueco idealmente ha de ser monoelemental para obtener la máxima intensidad de señal luminosa. Sin embargo, con lámparas multielementales pueden medirse concentraciones de 0,8μmol/L con una dilución del espécimen 1:1.
La línea que se utiliza es la de 324,7nm que está libre de interferencias y es la más intensa. La intensidad de corriente más habitual es de 15mA (25mA en las multielementales). La rendija se ajusta a 0,7nm y se orienta la lámpara de manera que la señal luminosa que llegue al detector sea máxima. Una vez realizado esto se ajusta la ganancia a 100 (ajuste automático en los aparatos actuales). En general, no es necesario un precalentamiento prolongado de la lámpara ya que los aparatos actuales compensan automáticamente las diferencias en intensidad de la emisión aunque un periodo de 10 a 15 min es recomendable.
Se emplea una llama de acetileno-aire con presiones comprendidas entre 85 y 100kPa, y 345 y 450kPa, respectivamente. El mechero habitual de 10cm de paso óptico es adecuado y ha de orientarse de manera que se obtenga la máxima señal con una solución acuosa de cobre de 31,5μmol/L mediante el ajuste en altura, profundidad y alineación. El haz de luz proveniente de la lámpara de cátodo hueco ha de formar un área nítida en el centro del mechero a 1cm por encima del mismo. Esto puede comprobarse interponiendo en el haz una hoja de papel blanco. La velocidad de aspiración del nebulizador adecuada es de 4mL/min y el tiempo de integración de la señal de, al menos, 0,5 s para evitar una oscilación demasiado elevada de la misma. Antes de realizar la lectura, dejar que se estabilice la señal (en los aparatos modernos esto se realiza automáticamente).
Otras comprobaciones adicionales que han de realizarse son: que exista agua en el bucle de seguridad del desagüe de la cámara de nebulización y que se encuentre anclado el mechero.
Preparación de la muestraDebido a la poca intensidad relativa de la línea de 324,7nm del cobre, la dilución de la muestra ha de ser la menor posible. Ha de llegarse a una solución de compromiso entre sensibilidad analítica y dilución que evite problemas de atasco en el tubo de aspiración del nebulizador y de partículas sólidas en la ranura del mechero. Se han propuesto diluciones 1:1 y 1:435. Con la primera se obtiene más sensibilidad analítica mientras que con la segunda (que es la empleada en el análisis del cinc) se evitan problemas de obstrucción. Asimismo, la dilución 1:1 permite detectar concentraciones muy bajas de cobre y el empleo de lámparas multielementales. Para evitar la obstrucción del nebulizador es suficiente emplear sueros bien desfibrinados y hacer un lavado con agua entre muestra y muestra de suero.
Preparación de los patronesEs suficiente preparar 3 soluciones patrón: 4,0, 8,0 y 16,0μmol/L (ya teniendo en cuenta que las muestras se introducen diluidas 1:1, con lo que los ajustes de la concentración han de programarse como 8,0, 16,0 y 32,0μmol/L). Estos patrones pueden prepararse con HCl 0,1mol/L34 o HNO3, aunque no es imprescindible. Para evitar diferencias de viscosidad con el suero se podría preparar con glicerina 0,66mol/L. Existen disoluciones comerciales de 15,80mmol/L a partir de la cual se puede preparar una solución de almacenamiento de 158μmol/L que es estable, al menos, 3 meses a temperatura ambiente. A partir de esta se preparan los patrones cada vez que se realicen las medidas.
El blanco se puede realizar con la solución de glicerina 0,66mol/L. Con esta se establece la línea base.
Características metrológicasEl procedimiento es lineal, al menos, hasta 31,5μmol/L. Presenta una imprecisión intraserial de alrededor del 3%, tanto en concentraciones en el límite inferior del intervalo de referencia como dentro del mismo o en la zona superior. La imprecisión interdiario y la inexactitud relativa son inferiores al 10%. Límite de detección: 0,6μmol/L. Este método es el de elección debido a su rapidez (ya que la preparación de los especímenes es mínima), economía de reactivos y relativa ausencia de interferencias.
Procedimiento de preparación del pelo para la medida de su contenido de cobreSe recomienda cortar unos trozos de unos 0,5cm de largo, del diámetro de un lápiz (aproximadamente 7mm) preferiblemente de la zona occipital y se lava con 5mL de Triton X100®, 10mL/L, mezclando durante 2 h. Después se enjuaga con agua desionizada varias veces y se añaden 3mL de EDTA-Na2 0,1mol/L agitando durante 2 h. Se vuelve a enjuagar con agua desionizada y se seca hasta peso constante a 75°C en un horno. Unos 50mg (mínimo 20mg) de pelo se digieren con 1mL de H2O2 10mol/L a 65-70°C hasta sequedad (unas 8 h). Se repite el proceso otra vez. El residuo se disuelve en 1mL de HNO3 0,5mol/L durante 2 h a temperatura ambiente. Posteriormente, se añaden 4mL de agua desionizada. Se mide por FAAS de manera análoga al suero (preparando los patrones en HNO3, 0,1mol/L) o bien mediante atomización electrotérmica o ICP-MS. Existen otros métodos estandarizados de preparación de la muestra de pelo, como el de la Agencia Internacional de Energía Atómica (IAEA).
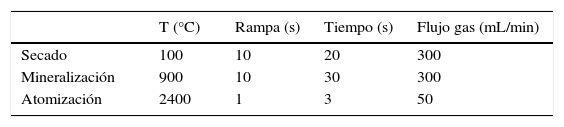
Procedimiento de medida de la concentración de cobre en orina por espectrometría de absorción atómica con atomización electrotérmicaCondiciones analíticasHa de emplearse una lámpara de cátodo hueco monoelemental para obtener la mayor intensidad de señal luminosa, la longitud de onda de 324,7nm y la rendija de 0,7nm. Las cámaras de grafito han de ser pirolíticas con plataforma tipo L’Vov. Existe un gran número de programas para el análisis de cobre. En general, suelen presentar una etapa, sencilla o doble, de secado (a 120°C o a 90 y 120°C, respectivamente), una de mineralización alrededor de 1.000°C y la de atomización alrededor de 2.300°C. Un programa rápido y al alcance de cualquier equipo es el representado en la tabla 1. Se recomienda utilizar un volumen de muestra de 20 μL. Idealmente, se emplea lectura en área de pico y la integración se lleva a cabo durante 4 o 5 s. Se puede añadir un paso adicional de limpiado a 2.600°C.
Preparación de la muestraEn general, es adecuada una dilución 1:5 de las muestras de orina, aunque también pueden emplearse diluciones 1:1. No hay definido ningún modificador de matriz que mejore la medición de la concentración de cobre. Puede usarse una solución de Mg(NO3)2 5,5mmol/L en Triton X-100® 2mL/L, o bien una solución solo con Triton X-100®.
Preparación de los patronesSe pueden preparar patrones de 0,4, 0,8 y 1,6mmol/L que se diluyen también 1:5 con el modificador de matriz de las muestras. A partir de una solución comercial de 15,8 mmol/L (p. ej., de la empresa Merck, referencia 1.09987.0001) se prepara una solución de almacenamiento de 158μmol/L en HNO3 0,1mol/L. Esta solución es estable, al menos, 3 meses a temperatura ambiente. Los patrones han de preparase en cada análisis. Tanto las puntas de pipeta como las cubetas y los contenedores han de estar lavados previamente con ácido y enjuagados con agua desionizada como en el caso del suero. El blanco consiste en una mezcla 1:5 de agua con modificador de matriz.
Características metrológicasEste procedimiento tiene una imprecisión intraserial inferior al 10% e interdiaria inferior al 15%. El límite de detección es de 0,1μmol/L. La AAS con atomización electrotérmica es la técnica de elección para el análisis de orinas debido a su mayor sensibilidad analítica (si se utiliza llama no se pueden separar adecuadamente los valores fisiológicos y los bajos, aunque generalmente lo que interesan son las concentraciones elevadas). Sin embargo, es una técnica relativamente lenta y necesita de un aparataje costoso y un cierto entrenamiento para su correcto empleo.
Procedimiento de medida de la concentración de cobre en diferentes muestras por espectrometría de masas con inducción acoplada de plasmaCondiciones analíticasA pesar de que las condiciones analíticas dependen del equipo utilizado, uno de los estándares interno más habitualmente empleado es el germanio, dado que posee una masa atómica muy similar a la del elemento a analizar cobre (masa atómica 72,64).
Preparación de las muestrasPara las muestras de suero y orina, se obtienen resultados satisfactorios utilizando diluciones 1:20 para suero de 1:10 para orina. El diluyente de suero/orina podría ser HNO3 suprapuro 5mL/L, etanol 10mL/L y Tritón X100 0,2mL/L para las muestras, los patrones y los controles.
Para las muestras de pelo (véase el apartado 4.4), una dilución 1:10 sería adecuada y el diluyente HNO3 suprapuro 10ml/L.
Preparación de patrones- a.
Suero. A partir de un patrón certificado de 1g/L de cobre (15,74mmol/L) se prepara una disolución acuosa intermedia y por disoluciones seriadas los patrones de la curva de calibración. También se podría preparar la disolución intermedia a partir de una solución comercial (p. ej., Merk de 30 elementos). Serían recomendables concentraciones de 2.000μg/L (31,47μmol/L), 1.000μg/L (15,74μmol/L), 500μg/L (7,87μmol/L), 100μg/L (1,57μmol/L) y 50μg/L (0,79μmol/L). Los patrones han de prepararse en cada análisis. Tanto las puntas de pipeta como las cubetas y los contenedores han de estar lavados previamente con ácido y enjuagados con agua desionizada.
- b.
Orina. Se puede preparar una disolución acuosa intermedia, bien a partir de un patrón certificado de 1g/L de cobre (15,74mmol/L) y por diluciones seriadas los patrones de la curva de calibración, o bien a partir de una solución comercial (p. ej., Merk de 30 elementos). Serían recomendables concentraciones de 1.000μg/L (15,74μmol/L), 500μg/L (7,87μmol/L), 100μg/L (1,57μmol/L) y 50μg/L (0,79μmol/L). Los patrones han de preparase en cada análisis. Tanto las puntas de pipeta como las cubetas y los contenedores han de estar lavados previamente con ácido y enjuagados con agua desionizada.
- c.
Pelo. Se puede preparar una disolución acuosa intermedia, bien a partir de un patrón certificado de 1g/L de cobre (15,74mmol/L) y por diluciones seriadas los patrones de la curva de calibración, o bien a partir de una solución comercial (p. ej., Merk de 30 elementos). Serían recomendables concentraciones de 50μg/L (0,79μmol/L), 20μg/L (0,31μmol/L), 10μg/L (0,16μmol/L) y 2μg/L (0,03μmol/L). Los patrones han de prepararse en cada análisis. Tanto las puntas de pipeta como las cubetas y los contenedores han de estar lavados previamente con ácido y enjuagados con agua desionizada.
Con el fin de estandarizar las condiciones analíticas es preferible la medida en tejido seco. El tejido (generalmente procedente de una punción-aspiración con aguja fina) se seca, en un recipiente exento de elementos traza, a 90°C durante el tiempo suficiente hasta que se consigue masa constante. Si la masa está comprendida entre 2 y 15mg, se somete a hidrólisis ácida con 200μL de HNO3 ultrapuro en un recipiente de polipropileno de 3-5mL de capacidad, cerrado a rosca herméticamente, durante 2-5 h, según se trate de tejido blando o hueso, respectivamente2. Posteriormente, se deja enfriar el recipiente antes de abrirlo y se añade agua desionizada a la disolución final adecuada (p. ej., se pueden añadir 1,8mL para obtener un volumen final de 2mL). La calibración más adecuada se realiza con un material de referencia (p. ej., hígado bovino NIST, ref. 1577) o bien por el método de adición de patrón, pero este tiene la desventaja de su lentitud. El sistema de corrección de fondo por efecto Zeeman es preferible en este tipo de análisis, aunque puede emplearse la corrección de fondo con lámpara de deuterio.
El principal problema de la determinación de cobre hepático, por ejemplo en la enfermedad de Wilson, es que la distribución del cobre en el interior del hígado con frecuencia no es homogénea. Por ello, la concentración puede ser infravalorada debido al error de muestreo. Se pueden dar falsos negativos en especímenes escasos, o en aquellos que presentan extensa fibrosis y pocas células parenquimatosas16.
Los sistemas que emplean hidróxido de tetrametilamonio o la calcinación de la materia orgánica presentan problemas de imprecisión, laboriosidad y riesgos de contaminación. La hidrólisis con otros ácidos, como el perclórico, presentan problemas de ajuste del volumen final.
Control de calidadEspecificaciones de la calidad analítica basadas en la variabilidad biológicaLos criterios de variabilidad biológica pueden aplicarse adecuadamente en el caso del cobre para establecer recomendaciones de calidad analítica36.
Teniendo en cuenta que la variación biológica para el cobre intraindividual e interindividual es del 4,7 y el 13,6%, respectivamente, las especificaciones deseables deben ser: el coeficiente de variación analítico inferior al 2,4%, el error sistemático inferior al 3,6% y el error analítico total inferior al 7,5%36.
Materiales de controlLa incorporación de materiales de referencia al proceso analítico es básica para la evaluación de la calidad y tiene un papel principal en la trazabilidad de las mediciones. Los laboratorios deben utilizar, en su trabajo diario, controles de calidad internos con matriz similar a la de los especímenes (suero, sangre, orina, pelo) a fin de asegurar la fiabilidad de sus resultados. Por ello, para suero se recomiendan los controles Lyphochek® Whole Blood Metals Quality Control (BIO-RAD, QCNetT™), Seronorm Trace Elements Serum (Seronorm™ Trace Element Serum [Nycomed Pharma AS, Oslo, Noruega]) u otros similares.
En la matriz de orina se sugieren los siguientes: Lyphochek® Urine Metals Control (BIO-RAD, QCNet™), Seronorm Trace Elements Urine (Nycomed Pharma AS, Oslo, Noruega), ClinChek® Urine Controls, lyophilised for Trace Elements (RECIPE®) u otros similares.
Programas de evaluación externa de la calidadAsimismo, es conveniente la participación en Programas de Garantía Externa de la Calidad (EQAS) de intercomparación de laboratorios, con el fin de asegurar la exactitud, la precisión y la reproducibilidad de sus resultados, además de ser requisito que exige la Entidad Nacional de Acreditación (ENAC). Indispensable para aquellos laboratorios clínicos que pretendan acreditarse por la Norma UNE-EN ISO 15189:2013. Existen numerosos programas externos de evaluación de la calidad específicamente diseñados para elementos traza que incluyen el cobre.
La SEQC participa en la organización del Programa de Garantía Externa de Calidad de la Occupational and Environmental Laboratory Medicine (OELM) en la que participan Australia, Francia, Italia, España, Bélgica y los Países Bajos. Estos países comparten las muestras, la base de datos y el diseño de los informes con un enfoque federativo, aunque se mantienen como organizaciones independientes con sus propias inscripciones al programa, idioma, reuniones de usuarios y otros sistemas de soporte (http://www.trace-elements.eu). Las matrices que presentan son suero, sangre y orina.
Otros programas a destacar son: el UKNEQAS TEQAS de Surrey (Inglaterra), que cuenta con muestras de suero, sangre, orina, y muestras sólidas, y el CTQ/INSPQ, del Centre de Toxicologie du Québec (Canadá), que dispone de las matrices suero, sangre, orina y pelo.
Intervalos de referenciaLa Comisión de Elementos Traza ha publicado el documento titulado «Valores de referencia del cobre en diferentes matrices. Revisión (2013)», en el que se realiza una recomendación sobre los valores de referencia a utilizar por los profesionales del laboratorio37. En dicho documento se insiste en la importancia de establecer y revisar periódicamente los valores/intervalos de referencia con el fin de garantizar y asegurar la calidad de los resultados ofrecidos.
Según esta revisión, se recomienda tomar como valores de referencia los siguientes:
- –
Suero/plasma: 700-1550μg/L (11,02-24,39μmol/L).
- –
Orina: < 40μg/día (< 0,63μmol/día)38,39.
- –
Biopsia hepática: 0-35μg/g peso seco (enfermedad de Wilson:>250μg/g) (> 3,93μmol/g).
Los autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Composición de la comisión: A. Anadón Ruíz, M. J. Ariza Astolfi, J. A. Cocho de Juan, P. Bermejo Barrera, M. J. Gaspar Blázquez, M. González Estecha, J. González Revaldería, S. Izquierdo Álvarez, M. T. Llorente Ballesteros (Presidenta desde 2012), J. L. López Colón, I. Palazón Bru, S. Pérez San Martín y E. Urrechaga Igartua
Los nombres de los componentes de la Comisión de Elementos Traza están relacionados en el anexo 1.
Estado de revisión: se trata de una actualización que sustituye a la anterior versión del documento de: González Revaldería J, Cocho de Juan JA. Metodología recomendada para la medición del contenido de cobre en especímenes biológicos. Documento A, Fase 3, Versión 4. Química Clínica. 2002;21(2):62-66. Entre los cambios más importantes realizados respecto de la versión anterior se encuentran una revisión de las nuevas técnicas de diagnóstico incluyendo tecnologías analíticas como la espectrometría de masas con plasma de acoplamiento inductivo (ICP-MS), actualización e inclusión de diferentes especímenes, su obtención y preparación, y, actualización del control de calidad y sus especificaciones, así como, inclusión de los intervalos de referencia actuales.

www.publicationethics.org.