


El déficit de carnitina palmitoiltransferasa II (CPT-II) se debe a mutaciones en el gen CPT2 y se asocia con 3 fenotipos. La forma adulta o muscular con crisis de mialgia y mioglobinuria es la más frecuente. Los fenotipos infantil y neonatal son multiorgánicos y de mayor gravedad. La mutación común en la forma adulta, p.S113L, no se ha descrito en casos de la variante neonatal. Se presenta un caso de forma adulta con mutaciones que previamente se han asociado a fenotipos diferentes.
Paciente y métodosPaciente de 22 años con episodios recurrentes de calambres musculares y orinas oscuras tras realizar esfuerzos prolongados de moderada intensidad, con una marcada elevación sérica de srm-creatincinasa (8.400U/l) y mioglobina (2.800ng/ml) y cuyo tejido muscular no mostró signos de miopatía. La actividad de CPT-II muscular se valoró radioquímicamente y el gen CPT2 se amplificó y secuenció en el secuenciador ABIPrism 310 (Applied Biosystems, Foster City, CA). La mutación p.R151Q se confirmó por reacción en cadena de la polimerasa (PCR)-fragmentos de restricción de longitud polimórfica (RFLP).
ResultadosLa actividad de CPT-II fue del 16% respecto del valor inferior de referencia. Se identificaron 2 mutaciones en heterocigosis en el gen CPT2: p.S113L y p.R151Q.
DiscusiónLa mutación p.R151Q únicamente se ha descrito en homocigosis y en heterocigosis compuesta (p.R151Q y p.P227L) en formas graves, y previamente no se ha asociado p.S113L a una forma clínica grave, lo que sugiere que la expresión del alelo p.S113L podría compensar los efectos deletéreos de la expresión del alelo p.R151Q, dando lugar al fenotipo moderado observado en la paciente.
Mutations in the CPT2 gene cause carnitine palmitoyltransferase (CPT-II) deficiency, which has been associated with three main phenotypes. The most frequent adult muscular form is characterized by recurrent episodes of myalgia and myoglobinuria. The infantile and neonatal variants are severe, multiorgan diseases. The commonest mutation, in the adult form, pS113L, has not been reported so far in neonatal cases. We report on an adult patient presenting with exercise intolerance who harboured mutations previously associated with diverse phenotypes.
Patient and methodsA 22 year-old woman presented with recurrent episodes of muscle cramps and dark urine after prolonged exercise of moderate intensity. She showed elevated serum CK (8400U/L) and myoglobin (2800ng/mL) levels. Muscle biopsy did not reveal signs of myopathy. Muscle CPT-II enzyme activity was determined by a radiochemical method. CPT2 gene was amplified and sequenced in an ABIPrism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). The p.R151Q mutation was confirmed by PCR-RFLP analysis.
ResultsThe activity of CPT-II was decreased (16% of the reference lower limit). Two heterozygous missense mutations were identified in the CPT2 gene: p.S113L and p.R151Q.
DiscussionThe p.R151Q mutation has been described in homozygous and compound heterozygous (p.R151Q and p.P227L) patients with the severe form. The p.S113L mutation has not been associated with “severe mutations”. We suggest that expression of the “benign” p.S113L allele might counteract the deleterious effects of the p.R151Q allele, which may account for the milder phenotype observed in the patient.
La intolerancia al ejercicio es un síntoma frecuente de consulta neurológica. Su etiología es multifactorial pues en la mayoría de las ocasiones forma parte de un cuadro sindrómico más complejo. No obstante, hay miopatías metabólicas que cursan con intolerancia al ejercicio como principal síntoma y cuya etiología bioquímica o genética no se llega a identificar en muchos pacientes, y las mejor estudiadas son algunas deficiencias enzimáticas que intervienen en la generación del ATP celular. Entre ellas destacan las deficiencias del metabolismo glucogenolítico y glucolítico1, como el déficit de la miofosforilasa (enfermedad de McArdle) que es la más frecuente de todas ellas2, el déficit del metabolismo purínico (mioadenilato desaminasa)3 y las alteraciones del metabolismo oxidativo mitocondrial (deficiencias de cadena respiratoria mitocondrial y mutaciones en ADN mitocondrial)4 y déficit del metabolismo lipídico, como el déficit de deshidrogenasa de ácidos grasos de cadena larga5. Entre estos últimos se encuentra el déficit de la carnitina palmitoiltransferasa (CPT-II)6. Algunos datos clínicos, como el tipo de ejercicio que desencadena los síntomas, y de laboratorio, como los valores de srm-creatincinasa (CK) en reposo, así como la medida de los valores de srm-lactato y srm-amonio ión en la prueba de ejercicio en isquemia en el antebrazo permiten delinear el diagnóstico clínico diferencial1. Otras pruebas como los tests ergométrico-fisiológicos pueden ayudar al diagnóstico7,8. Al diagnóstico definitivo se llega demostrando el déficit enzimático en tejido muscular o identificando mutaciones en los genes correspondientes en sangre o músculo9.
La CPT-II es una proteína homotetramérica que se encuentra en la membrana interna mitocondrial y que facilita el transporte de los ácidos grasos de cadena larga al interior de la matriz mitocondrial junto a la acción de la CPT-I y la acil carnitina translocasa10. Es una proteína ubicua y únicamente presenta una isoforma codificada por el gen CPT2 (cromosoma 1p32) que está compuesto por 5 exones11.
La deficiencia muscular de esta enzima es la alteración más frecuente del metabolismo lipídico. Sin embargo, se han descrito 3 fenotipos para la deficiencia de la CPT-II. La forma más prevalente (se han descrito más de 200 casos) es el fenotipo adulto o muscular (MIM 255110)12 que cursa con ataques recurrentes de mialgia y frecuentes episodios de mioglobinuria cuyos agentes precipitantes son el ejercicio físico, el frío, la fiebre o el estrés. Es de destacar que los valores de CK en reposo suelen ser normales. Las otras 2 presentaciones son más raras pero más graves: a) el fenotipo infantil o hepatocardiomuscular (del que se han descrito 16 casos ) (MIM:600649)13 cuya sintomatología aparece entre los 6 meses y los 2 años de vida, que se caracteriza por hipocetosis, hipoglucemia con afectación multiorgánica llegando a producir fracaso hepático, cardiomiopatía y miopatía periférica, falleciendo la mayoría de los pacientes de forma repentina durante su infancia debido a la afectación cardíaca, y b) el fenotipo neonatal (MIM 608836) cuya presentación es similar a la forma infantil pero acompañada de malformaciones y que se inicia desde pocas horas a 4 días posteriores al nacimiento y es letal durante el primer mes de vida13.
El diagnóstico bioquímico y molecular de esta deficiencia se realiza mediante un análisis enzimorradiométrico que demuestra la deficiencia de la actividad enzimática de la CPT-II en músculo o fibroblastos14, y/o mediante análisis molecular de mutaciones en el gen CPT211. En este sentido, cabe decir que aunque se han descrito más de 60 mutaciones patogénicas, alrededor del 60%15,16 de los pacientes con fenotipo muscular presenta la mutación p.S113L. Por otro lado, se ha logrado establecer, en cierta medida, algún tipo de asociación para algunas mutaciones con cada uno de los 3 fenotipos descritos9,17.
En este artículo se presenta un caso de forma adulta con mutaciones que previamente se han asociado a fenotipos diferentes.
Paciente y métodosPacienteMujer de 22 años, sin historia familiar de miopatía ni de enfermedad genética que trabajó como monitora de aeróbic durante el año anterior, que inicia, en un contexto de cuadro febril, con dolor intenso y progresivo en los miembros inferiores mientras ejercía de camarera. Al ingresó en urgencias el electrocardiograma, la imagen radiográfica de tórax y el electromiograma fueron normales. Las pruebas de laboratorio mostraron unos valores de CK de 8.400Ul (valor de referencia [VR]<170), srm-mioglobina de 2800ng/ml (VR<74) y srm-creatininia de 0,85mg/dl (VR<1,1). Presentó una mioglobinuria intensa. El test de drogas de abuso en orina resultó negativo. Se practicó una biopsia muscular en la que los hallazgos morfológicos no fueron relevantes y no se observaron acúmulos de lípidos ni de glucógeno. Las tinciones histoquímicas demostraron la presencia de actividades de enzimas relacionadas con otras deficiencias que producen intolerancia al ejercicio, como miofosforilasa, fosfofructocinasa y mioadenilato deaminasa.
Métodos- –
Análisis bioquímico. La actividad de la CPT-II se midió en homogenado de músculo esquelético mediante el método de intercambio isotópico de Norum14 con adición de 300nM de palmitoil-carnitina
- –
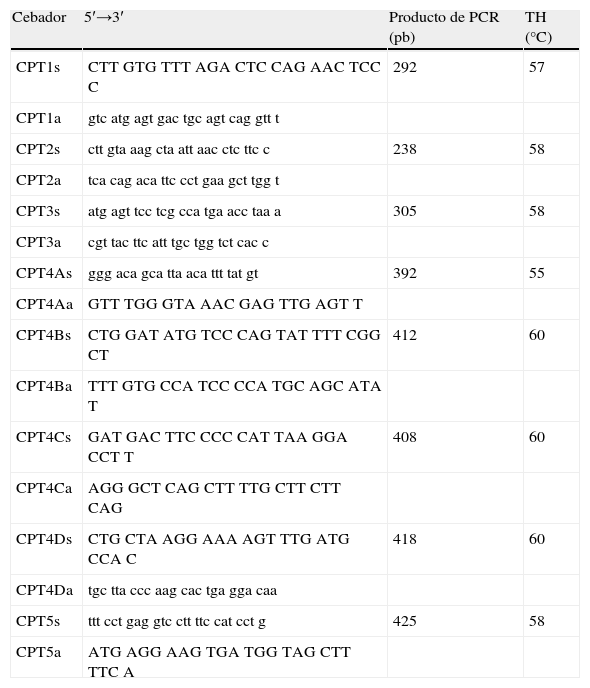
Análisis genético-molecular. El ADN de músculo se aisló con un método estándar basado en la extracción con fenol-cloroformo tras digestión con proteinasa K recombinante PCR Grade (Roche Diagnostics, Mannheim, Germany). Se amplificó la región codificante y las regiones de unión intrón-exón del gen CPT2 mediante reacción en cadena de la ADN polimerasa (PCR) en 8 fragmentos, dado que el exón 4 se amplificó en 4 fragmentos solapados por su gran tamaño (1.305pb) (tabla 1), que se purificaron con el kit Illustra™ GFX™ PCR ADN y Gel Band Purification Kit (GE Healthcare, Buckinghamshire, UK). Se secuenciaron ambas hebras del ADN amplificado siguiendo las instrucciones del fabricante del d-Rhodamine Cycle Sequencing Terminador Kit en un secuenciador ABIPrism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA.).
Tabla 1.Cebadores utilizados para amplificar la región codificante y las regiones adyacentes de las uniones intrón/exón del gen CPT2
Cebador 5′→3′ Producto de PCR (pb) TH (°C) CPT1s CTT GTG TTT AGA CTC CAG AAC TCC C 292 57 CPT1a gtc atg agt gac tgc agt cag gtt t CPT2s ctt gta aag cta att aac ctc ttc c 238 58 CPT2a tca cag aca ttc cct gaa gct tgg t CPT3s atg agt tcc tcg cca tga acc taa a 305 58 CPT3a cgt tac ttc att tgc tgg tct cac c CPT4As ggg aca gca tta aca ttt tat gt 392 55 CPT4Aa GTT TGG GTA AAC GAG TTG AGT T CPT4Bs CTG GAT ATG TCC CAG TAT TTT CGG CT 412 60 CPT4Ba TTT GTG CCA TCC CCA TGC AGC ATA T CPT4Cs GAT GAC TTC CCC CAT TAA GGA CCT T 408 60 CPT4Ca AGG GCT CAG CTT TTG CTT CTT CAG CPT4Ds CTG CTA AGG AAA AGT TTG ATG CCA C 418 60 CPT4Da tgc tta ccc aag cac tga gga caa CPT5s ttt cct gag gtc ctt ttc cat cct g 425 58 CPT5a ATG AGG AAG TGA TGG TAG CTT TTC A PCR: reacción en cadena de la polimerasa. “TH: temperatura de hibridación.
La mutación p.R151Q se confirmó mediante el diseño de un método de PCR- fragmentos de restricción de longitud polimórfica (PCR-RFLP) usando los cebadores (tabla 2) y las condiciones de amplificación y digestión con la enzima de restricción Ava I descritas previamente18.
ResultadosLa actividad enzimática de la carnitina palmitoiltransferasa II en homogenado muscular fue de 0,04nmol×min−1×mg proteína no colágena−1 (valor inferior de referencia, 0,25 (P2.5; n=30), representando un 16% de actividad.
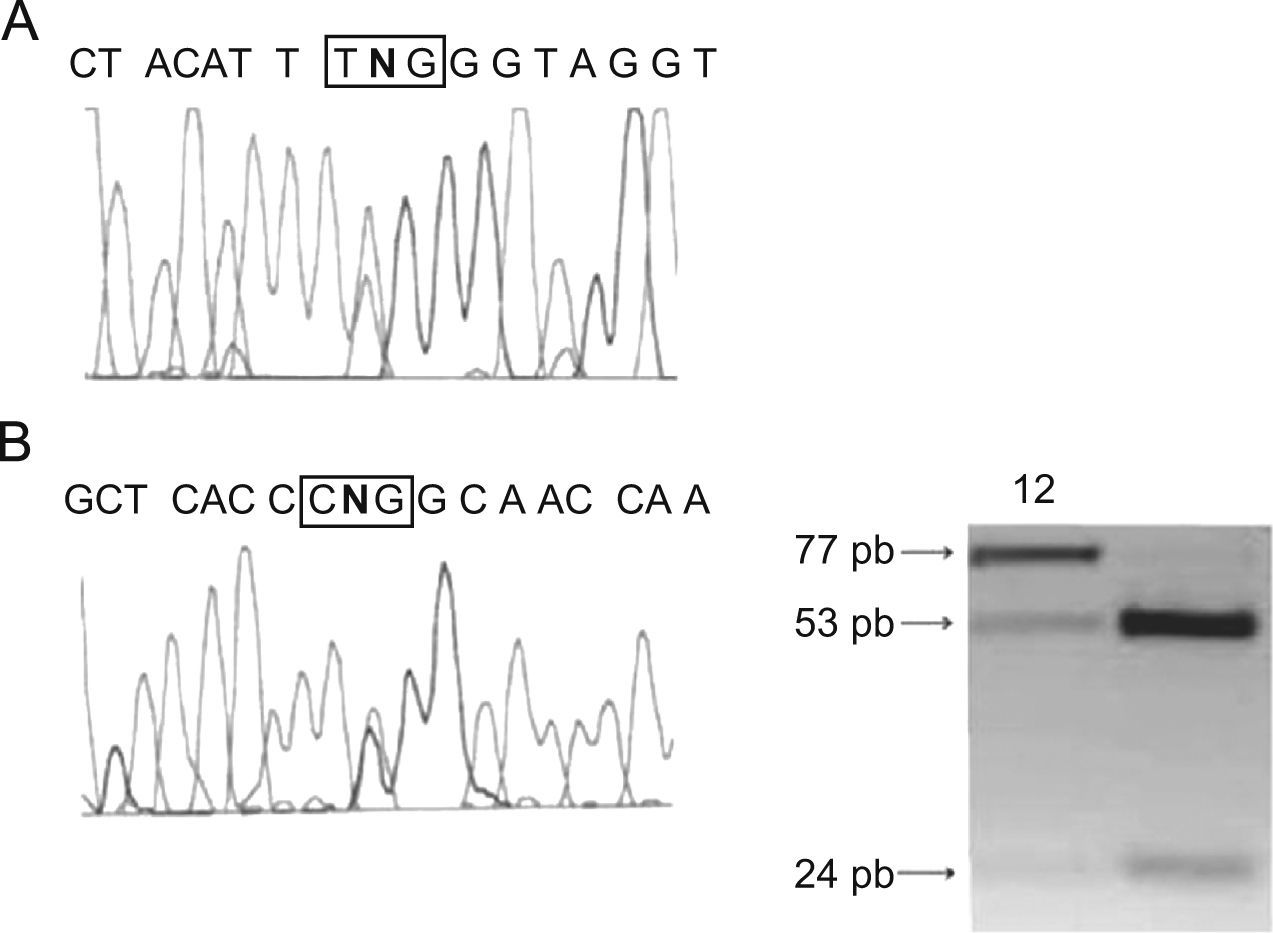
Mediante secuenciación del gen CPT2 en ADN aislado de músculo esquelético se identificaron 2 mutaciones de sentido equivocado, es decir mutaciones que predicen un cambio de aminoácido, en heterocigosis: la mutación común p.S113L en el exón 3 (fig. 1A) y otra mutación previamente descrita, p.R151Q en el exón 4 (fig. 1B). Esta última mutación se confirmó mediante PCR-RFLP (fig. 1B).
 Análisis por secuenciación de la mutación p.S113L (c.338C>T) en exón 3 del gen CPT2. El recuadro indica el codón 113 alterado. (B) Identificación de la mutación p.R151Q (c.452G>A) exón 4 del gen CPT2. lzquierda: el recuadro indica el triplete lesionado en la secuenciación del exón 4; derecha: análisis de PCR-RFLP en ADN muscular. Calle 1: paciente; calle 2: control. El amplicón de 77pb se digirió con la enzima de restricción AvaI que genera 2 fragmentos de 53pb y 24pb en presencia del alelo normal, mientras que el alelo mutado pierde el sitio de restricción permaneciendo intacto. PCR: reacción en cadena de la polimerasa; RFLP: fragmentos de restricción de longitud polimórfica.")
(A) Análisis por secuenciación de la mutación p.S113L (c.338C>T) en exón 3 del gen CPT2. El recuadro indica el codón 113 alterado. (B) Identificación de la mutación p.R151Q (c.452G>A) exón 4 del gen CPT2. lzquierda: el recuadro indica el triplete lesionado en la secuenciación del exón 4; derecha: análisis de PCR-RFLP en ADN muscular. Calle 1: paciente; calle 2: control. El amplicón de 77pb se digirió con la enzima de restricción AvaI que genera 2 fragmentos de 53pb y 24pb en presencia del alelo normal, mientras que el alelo mutado pierde el sitio de restricción permaneciendo intacto. PCR: reacción en cadena de la polimerasa; RFLP: fragmentos de restricción de longitud polimórfica.
Se ha descrito que hay cierta asociación entre cada uno de los 3 fenotipos de deficiencia de CPT II y la presencia de unas determinadas mutaciones13. Además, se ha propuesto que la CPT-II se puede asociar con componentes distales de la beta-oxidación, de manera que mutaciones en distintas posiciones de la proteína provocarían una eficiencia variable del sistema de oxidación, o bien afectarían en algún grado a la unión de la enzima a la membrana interna mitocondrial, provocando eficiencias variables de actividad13,19,20. Se ha observado que las mutaciones descritas para el fenotipo muscular se caracterizan porque pueden encontrarse en homocigosis (p.P50H, p.S113L y p.R161W) o en heterocigosis, existiendo siempre, en este caso, una mutación localizada en el exón 4 en heterocigosis compuesta con p.S113L (exón 3), la más frecuente del fenotipo adulto, o con p.P50H (exón 1). Sin embargo, las formas graves se han asociado con mutaciones que se localizan en los exones 4 y 5 (p.R151Q, p.P227L, p.D328G, p.F383Y) las cuales pueden encontrarse en homocigosis o en heterocigosis pero con la peculiaridad de que nunca se han descrito en asociación con las mutaciones p.S113L o p.P50H. Nuestro caso es una paciente de 22 años con fenotipo muscular en la que se identificó la presencia de 2 mutaciones, p.R151Q y p.S113L, cuya heterocigosis compuesta no se había descrito anteriormente. Además, la mutación p.R151Q (exón 4), únicamente se ha identificado en 2 casos, ambos asociados a un fenotipo grave. El primer caso18 es un fenotipo neonatal de heterocigosis compuesta para las mutaciones p.R151Q y p.P227L las cuales únicamente se han documentado hasta ahora en fenotipos graves. El otro paciente fue un caso de fenotipo grave que presentó la mutación p.R151Q en homocigosis10. El hecho de que estos 2 pacientes expresaran formas graves del déficit de CPT II, y que nuestra paciente con la mutación p.R151Q asociada con la mutación más frecuente del déficit en el adulto, p.S113L, expresara un fenotipo muscular, sugiere que la expresión del alelo p.S113L podría, de alguna manera, compensar los efectos deletéreos de la expresión del alelo p.R151Q ejerciendo de esta manera un efecto protector contra la aparición del fenotipo grave, como se ha indicado para otras mutaciones13.
Por otra parte, un ejemplo de las dificultades de establecer relaciones entre el genotipo y el fenotipo de este déficit es el hecho de que la mutación p.R631C se haya descrito en los 3 fenotipos con diferente expresión intrafenotipo21, lo que apunta a que podrían haber, además del genotipo, otros factores implicados en el desarrollo de la enfermedad, como factores ambientales, el efecto de diferentes polimorfismos (asociación de polimorfismos p.V368I y p.M647V con la mutación p.S113L)15, y el sexo, debido a que el fenotipo muscular de herencia autosómica recesiva se manifiesta en un mayor porcentaje en varones que en mujeres.
Recientemente, Corti et al22 han identificado en 2 familias que expresan clínicamente la forma muscular de déficit de CPT II la presencia de heterocigosis compuesta para las mutaciones p.R151Q y p.S113L. Estos hallazgos confirman que la mutación p.S113L puede jugar un papel modulador de la los efectos fenotípicos de la mutación p.R151Q, e indican que dicha alteración está presente en diferentes grupos poblacionales, lo que subraya su carácter patogénico.
La identificación molecular de las mutaciones responsables del déficit de CPT2 puede ayudar: a) al diagnóstico diferencial con otras IE pues no se dispone de una prueba histoquímica en músculo para el diagnóstico de este déficit, y su diagnóstico bioquímico depende de un método radiactivo; b) a comprender la compleja relación entre el genotipo y el rango de severidad de los fenotipos tan variables de este déficit, y c) puede tener importantes implicaciones en las predicciones fenotípicas en casos de diagnóstico prenatal.
Este trabajo corresponde a una comunicación científica presentada y premiada en el I Congreso Nacional del Laboratorio Clínico celebrado en Sevilla del 17 al 20 de octubre de 2007
Rosa Pello está financiada por el programa post-Formación Sanitaria CM05/00088 del Instituto de Salud Carlos III (ISCIII), Fondo de Investigación Sanitaria (FIS). Sara Jiménez está financiada por un contrato de Técnico de Laboratorio del programa de recursos humanos del CIBER de Enfermedades Raras, ISCIII. Miguel A. Martín es beneficiario en el año 2008 del Programa de Intensificación de la Actividad Investigadora, ISCIII-Agencia Laín Entralgo (Comunidad de Madrid). El trabajo ha sido realizado con fondos de los proyectos de investigación del FIS-ISCIII PI04/0467, FIS06/0547 y de la Fundación Mutua Madrileña 2005-59.

www.publicationethics.org.