


El linfoma es la forma más prevalente de neoplasia hematológica, clasificándose en dos grandes grupos, Hodgkin y no Hodgkin. El linfoma de células del manto (LCM) es un subtipo de linfoma no Hodgkin tipo B, de carácter agresivo y que tiene su origen en células de la zona periférica del centro germinal o en la zona del manto del folículo linfoide.
El LCM es uno de los linfomas menos frecuentes, suponiendo cerca del 7% de los casos de linfoma no Hodgkin en los EE. UU. y Europa, con una incidencia aproximada de 4 a 8 casos por millón de personas y año. En torno a tres cuartas partes de los pacientes son varones caucasianos, reduciéndose a la mitad la frecuencia en la raza negra. La edad media en el diagnóstico es de 68 años, desarrollando adenopatías en el 75% de los casos y produciéndose diseminación extranodal en el 25% restante (bazo, médula ósea, sangre, tracto gastrointestinal, mama, pleura y anejos oculares).
El laboratorio es un pilar clave en el diagnóstico de LCM establecido mediante el estudio histológico, inmunofenotípico y molecular.
El 70% de los pacientes que se diagnostican presentan la enfermedad en un estadio avanzado. Ello conlleva frecuentemente la extensión gastrointestinal y a médula ósea.
Aunque los linfomas afectan principalmente a ganglios linfáticos y tejidos linfoides, en algunos casos invaden la médula ósea y sangre periférica. Este proceso recibe el nombre de leucemización y ocurre en el 35% de los casos. Ante una cifra de leucocitos elevada en un paciente diagnosticado de LCM, el cometido del laboratorio será observar el frotis de sangre periférica en busca de linfocitos de aspecto centrocítico.
Con los tratamientos convencionales, la supervivencia media era de 3 años, cifra que ha aumentado hasta los 7 años para los pacientes que reciben los nuevos tratamientos. El pronóstico de LCM leucemizado es inferior al de las formas ganglionares sin expresión periférica.
Lymphoma is the most prevalent kind of hematopoietic neoplasm, and it can be classified into two groups, Hodgkin and non-Hodgkin lymphoma. Mantle cell lymphoma (MCL) is one of the mature B cell non-Hodgkin lymphomas, with aggressive behavior and originates in peripheral zone cells of the germinal center or in the mantle zone of lymphoid.
The mantle cell lymphoma is one of the less common lymphomas, and comprises about 7% of cases of non-Hodgkin lymphoma in the US and Europe, with an incidence of 4 to 8 cases per million persons per year. Around three-quarters of affected patients are Caucasian males, halving the rate in blacks. Median age at diagnosis is 68 years. Approximately 75% of patients initially present with lymphadenopathy and disease is the primary presentation in the remaining 25% (spleen, bone marrow, blood, gastrointestinal tract, breast, pleura and eye orbit).
The Laboratory is a key pillar in the diagnosis of MCL established by histological, immunophenotypic and molecular study.
Most patients with mantle cell lymphoma (70%) develope advanced stage disease at diagnosis, involving gastrointestinal tract and bone marrow.
Although patients mainly present with lymphadenopathy and other lymphoid tissues participation, sometimes lymphoma invades the bone marrow and peripheral blood. This process is called leukemization and occurs in 35% of cases. Faced with a very high number of leukocytes in a patient already diagnosed with MCL, the role of the laboratory will be to observe the smear blood for lymphocyte centroblastic appearance.
With conventional treatments median survival was 3 years, which has increased to 7 years with new treatments. Leukemized MCL prognosis is lower than in a ganglion forms without peripheral expression.
Varón de 67 años que presenta como antecedentes cólicos nefríticos, hipertrofia benigna de próstata y adenocarcinoma de colon transverso tratado con cirugía y quimioterapia adyuvante, quedando el paciente libre de enfermedad.
Tras realizar una tomografía axial computarizada de control, se observa una masa retroperitoneal. El estudio anatomopatológico concluye que se trata de una proliferación linfoide atípica de célula pequeña que se dispone con un patrón tipo manto, con conservación de los centros germinales.
El estudio inmunohistoquímico muestra positividad para ciclina D1, CD20 y CD5. Se trata de un linfoma de células del manto estadio de Ann Arbor IV A (IV por afección diseminada de localizaciones extralinfáticas con o sin afectación ganglionar, en este caso, con infiltración de médula ósea y A por ausencia de síntomas generales). MIPI (MCL International Prognostic Index) de riesgo bajo.
Se le administran 3 líneas de tratamiento con respuesta parcial, alternando periodos libres de enfermedad con recaídas (progresión ganglionar en primer lugar y esplénica y ósea en segundo lugar).
Posteriormente, el paciente acude a urgencias por cuadro de un mes evolución con empeoramiento en la última semana consistente en malestar general, debilidad generalizada con dificultad para la deambulación y fiebre. En la tomografía axial computarizada realizada pocos días antes en el servicio de oncología, se observa un crecimiento de adenopatías de forma significativa en cuello, tórax y abdomen, con afectación de asa de intestino delgado junto con esplenomegalia de nueva aparición, lo que concuerda con una recidiva de linfoma.
La hematimetría realizada en el Advia 2120i (Siemens) revela una cifra de leucocitos de 99,0×109/l (valor de referencia 4,2-11,5×109/l) con un valor absoluto de linfocitos de 75,9×109/l (valor de referencia 1,0-5,0×109/l) y trombocitopenia con una cifra de plaquetas de 33×10^9/l (valor de referencia 120-450×109/l). El autoanalizador utiliza la tinción de peroxidasa para diferenciar las células sanguíneas. Las células atípicas corresponden a las de tamaño grande y negativas para dicha tinción (células grandes no teñidas [large unstained cells –LUC–]). En esta subpoblación se engloban los linfocitos reactivos, las células plasmáticas y las células blásticas peroxidasa negativas (18,6% en nuestro caso).
Con estos datos, procedemos a observar el frotis sanguíneo.
DiscusiónEl recuento diferencial manual o estudio citológico de sangre periférica es actualmente una herramienta imprescindible para el diagnóstico. La sangre constituye un fluido orgánico fácilmente accesible, por lo que es el punto de partida más idóneo para el estudio de enfermedades y procesos hematológicos que tienen expresión periférica. Los criterios numéricos del hemograma para realizar la fórmula leucocitaria son: leucocitos superiores a 20×109/l, hematocrito inferior a 0,25 L/l, plaquetas inferiores a 50×109/l, linfocitos superiores al 60% o superiores a 5,5×109/l, monocitos superiores al 15%, eosinófilos superiores al 20%, basófilos superiores al 2,5% o valor de LUC superior al 5%1.
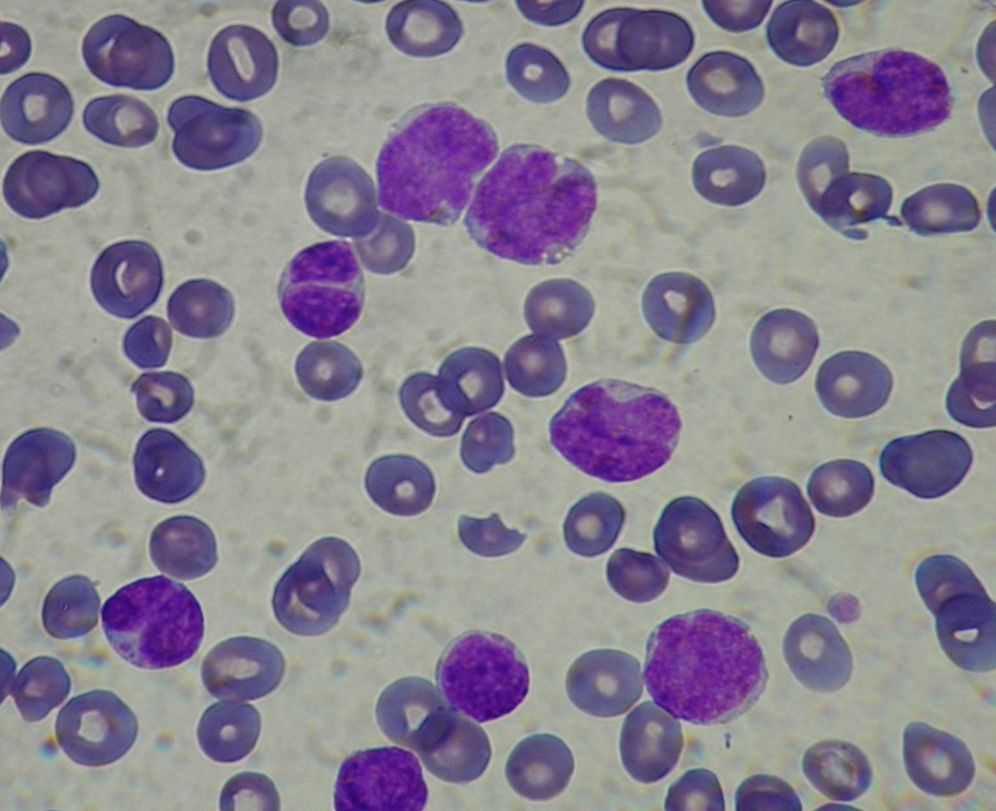
En la extensión de sangre periférica del paciente, se observa anisocitosis de la serie roja, eritroblastos aislados, y una población nucleada constituida en un 98% por linfocitos de morfología variada. La mayor parte de estos linfocitos tienen aspecto centrocítico (con hendidura central) junto con otros de mayor tamaño y cromatina laxa (fig. 1). También se observan algunas células blásticas. El porcentaje de neutrófilos es del 2%. Dicho pleomorfismo celular concuerda con el diagnóstico de linfoma de células del manto (LCM) leucemizado.

La diferencia entre los datos aportados en el hemograma realizado en el autoanalizador y el recuento hecho manualmente se debe a la dificultad del equipo para clasificar esta población nucleada con una morfología celular alterada.
Aunque se estima que este proceso ocurre en el 35% de los casos de LCM, la frecuencia exacta de un cuadro leucémico en este tipo de linfoma, bien en el diagnóstico o durante su evolución, no se conoce con exactitud ya que una proporción sustancial de casos se diagnostican como leucemias linfocíticas crónicas atípicas.
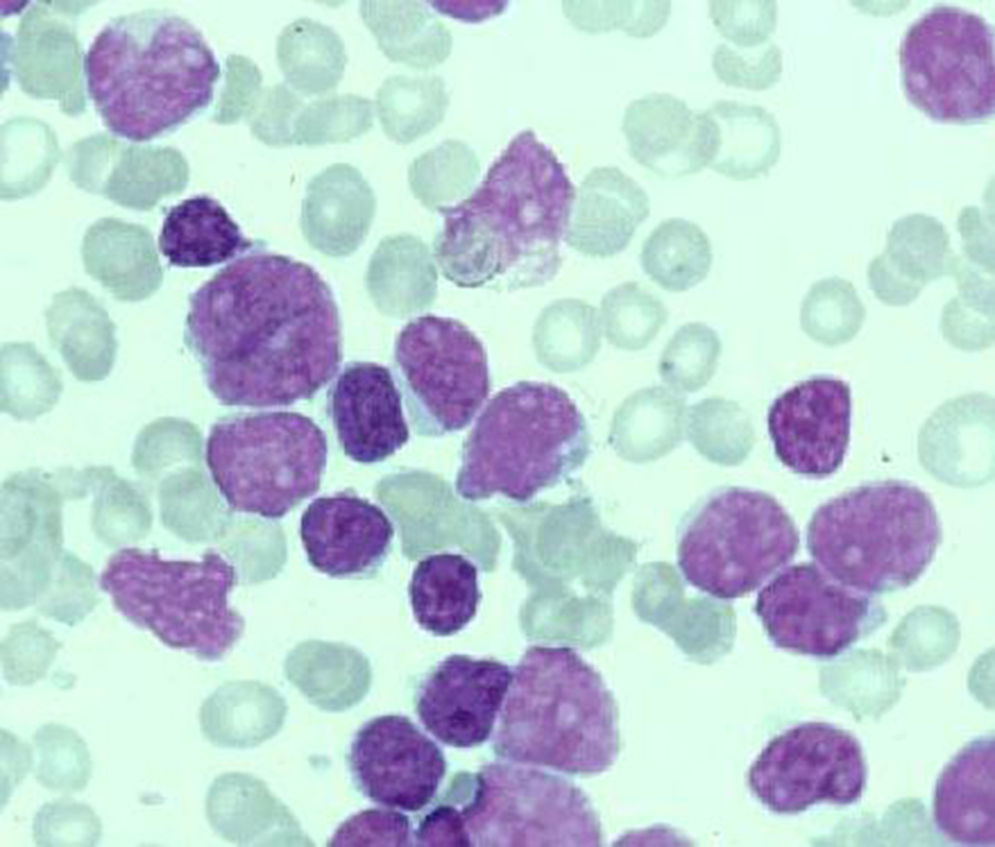
El cuadro morfológico en sangre periférica en el LCM se caracteriza por un marcado pleomorfismo en lo que se refiere a tamaño celular e irregularidad del contorno nuclear. La célula más típica es un linfocito de tamaño medio con un núcleo de cromatina punteada, una o dos hendiduras de corta longitud y un citoplasma escaso o mediano; el nucléolo no es visible o en caso de serlo es pequeño y no prominente2,3. Actualmente se considera que el LCM puede transformarse a formas anaplásicas y/o blásticas (fig. 2) lo que le confiere un peor pronóstico a un linfoma que, de por sí, es de difícil manejo clínico por su resistencia al tratamiento o respuesta corta al mismo.

El diagnóstico diferencial de LCM leucemizado se plantea con la leucemia linfocítica crónica ya que esta suele manifestar un cierto pleomorfismo, con la leucemia prolinfocítica B, especialmente en casos asociados a la traslocación cromosómica t(11;14) y con otros linfomas leucemizados, en particular el folicular y el esplénico de células vellosas.
Además de la citología, la histología es un pilar esencial para el diagnóstico diferencial entre estas entidades y los marcadores inmunológicos y la citogenética son asimismo de gran valor siempre que se tenga presente que la t(11;14) observada del 40-70% de los casos mediante citogenética convencional y en el 95% mediante hibridación in situ fluorescente no es estrictamente específica de LCM.
Respecto al inmunofenotipo, las células del linfoma del manto expresan en su superficie gran cantidad de IgM e IgD, además de CD5 y FMC74,5. Cuando existe extensión gastrointestinal (poliposis linfomatosa) las células tumorales también expresan CD49d6,7. La tinción nuclear para la proteína ciclina D1 es positiva en el 95% de los casos8,9. Esta sobreexpresión se asocia a la traslocación cromosómica t(11;14)(q13;q32), que afecta a los genes BCL1, CCND1, PRAD1 y al gen de las cadenas pesadas de las inmunoglobulinas (IgH)10,11.
ConclusionesSe describe un caso clínico de un paciente con leucemización de LCM. Se trata de una patología más frecuente en hombres que en mujeres (proporción 4:1) con una edad de aparición próxima a los 65 años12.
Los síntomas más frecuentes son fiebre, sudoración nocturna, y pérdida de peso. Las adenomegalias y esplenomegalia suelen estar presentes.
Para el diagnóstico definitivo, se requiere el análisis histológico de biopsias.
En el estudio de extensión existen pruebas complementarias como tomografía axial computarizada, tomografía por emisión de positrones, colonoscopia o endoscopia. En pacientes con variante blástica, se evalúa el líquido espinal.
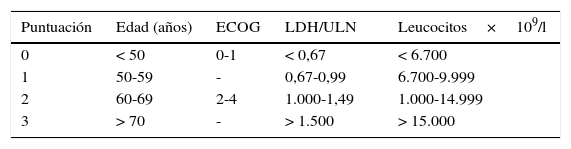
El pronóstico de LCM es complejo, por ello recientemente se diseñó un índice pronóstico específico (MIPI)13–15. Dicho índice tiene en cuenta la edad, el estado funcional, el valor de la lactatodeshidrogenasa (400 U/l en nuestro paciente) y el recuento de leucocitos en el momento del diagnóstico, estratificando a los pacientes en 3 grupos de riesgo; bajo, medio o alto. Existe una modificación simplificada del MIPI (tabla 1) que no contempla el índice de proliferación celular Ki67. Mediante el estudio de la enfermedad mínima residual se puede evaluar si la respuesta al tratamiento ha sido completa, es decir, si la enfermedad persiste bajo el nivel de detectabilidad de las técnicas convencionales. La enfermedad mínima residual en el LCM se estudia mediante la determinación del reordenamiento BCL-1 por técnicas de reacción en cadena de la polimerasa.
El tratamiento más común es la inmunoterapia con quimioterapia seguida o no de trasplante de médula ósea, e incluso radioterapia.
El LCM es una enfermedad sistémica con frecuente implicación de la médula y del aparato gastrointestinal, y puede existir una fase leucémica con presencia de linfocitos característicos en sangre. La observación de leucocitosis y trombocitopenia, junto con aumento de las LUC en un hemograma, sugiere que probablemente el paciente presente una enfermedad hematológica. Por tanto, el laboratorio tiene un papel primordial no solo en la observación morfológica de dichas muestras, sino en la búsqueda de la alteración genética desencadenante de la enfermedad y en la determinación de parámetros como la lactatodeshidrogenasa con el objeto de calcular el pronóstico de la enfermedad.
FinanciaciónNo se ha recibido financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Quiero agradecer a los autores y a todo el personal del Área de Diagnóstico Biológico del Hospital Universitario de la Ribera su inestimable colaboración.
