




La enfermedad de von Hippel Lindau (VHL) (OMIM: 193300) es un síndrome familiar de predisposición al cáncer, asociado a una variedad de tumores benignos y malignos, principalmente hemangioblastomas en retina y en el sistema nervioso central, carcinoma de células renales y feocromocitomas.
Exponemos el caso de un niño de 8 años con hipertensión arterial y antecedentes familiares de feocromocitoma, que acude a Urgencias por presentar visión borrosa. Se observa en la ecografía-doppler abdominal una masa suprarrenal izquierda y en la analítica una elevación de los niveles de normetanefrinas en orina. La identificación de una mutación en el gen VHL (OMIM: 608537) confirmó el diagnóstico de enfermedad VHL. Debe sospecharse el diagnóstico en pacientes con feocromocitoma de aparición precoz, más aún con antecedentes familiares de este tipo de tumores.
Von Hippel–Lindau (VHL) disease (OMIM: 193300) is a familial cancer syndrome, associated with various benign and malignant tumours, mainly retinal and central nervous system haemangioblastomas, renal cell carcinomas and pheochromocytomas.
We describe the case of a 8 years old patient with arterial hypertension, blurred vision and family history of pheochromocytoma. A left adrenal mass is observed in doppler ultrasonography and high levels of normetanephrines in orine. Identification of a mutation in the VHL gen (OMIM: 608537) confirms the diagnosis of VHL disease. The diagnosis should be suspected in patients with early onset of pheochromocytoma, even more if there is a family history of this kind of tumors.
La enfermedad de von Hippel Lindau (VHL) es un trastorno genético de herencia autosómica dominante causado por la inactivación del gen supresor de tumores conocido como VHL (OMIM: 608537), localizado en la región cromosómica 3p25-26. Mutaciones en este gen están asociadas al desarrollo de tumores en diferentes órganos. La incidencia es baja, aproximadamente de 1/36.000 nacidos/año, por lo que se considera una enfermedad rara o poco frecuente1.
Los pacientes afectados muestran mayor predisposición a desarrollar hemangioblastomas en retina y en el sistema nervioso central (SNC), feocromocitomas, carcinomas de células renales, quistes renales y pancreáticos, tumores del saco endolinfático, cistoadenomas benignos del epidídimo en varones y tumores del ligamento ancho en mujeres1,2.
Las familias con VHL se pueden clasificar en función de la presentación clínica en tipo 1 y tipo 2. A su vez el tipo 2 se subdivide en 2A, 2B y 2C (tabla 1). Todos los tipos están asociados a feocromocitoma, excepto el tipo 1. Los hemangioblastomas tanto de retina como de SNC se desarrollan en los tipos 1, 2A y 2B. Los pacientes clasificados en el tipo 1 o en el tipo 2B pueden presentar cáncer renal, que es lo que determina el pronóstico; también asocian tumores y quistes pancreáticos en estos 2 tipos. En el tipo 2C solo se desarrolla feocromocitoma como único signo de VHL3.
Clasificación de las familias VHL de acuerdo con la presentación clínica
| Tipo 1 | Angiomas de retina (hemangioblastomas) Hemangioblastomas en el sistema nervioso central Cáncer renal Tumores y quistes pancreáticos |
| Tipo 2A | Feocromocitomas Hemangioblastomas de retina Hemangioblastomas del sistema nervioso central |
| Tipo 2B | Feocromocitomas Hemangioblastomas de retina Hemangioblastomas del sistema nervioso central Cáncer renal Tumores y quistes pancreáticos |
| Tipo 2C | Solo feocromocitomas |
La enfermedad VHL presenta alto grado de penetrancia (80–90%) pero con gran variabilidad fenotípica. No está muy clara la correlación entre los genotipos específicos de VHL y las expresiones fenotípicas. Sin embargo, ante un paciente diagnosticado con feocromocitoma debe considerarse su estudio genético, más aún en casos con historia familiar previa, porque este tipo de tumores suele asociarse a síndromes genéticos. Por ello, cuando se diagnostica la enfermedad de VHL en un paciente y en sus familiares, es necesario un seguimiento clínico estrecho4.
Presentación del casoPaciente de 8 años que acude a Urgencias por presentar visión borrosa de 48 h de evolución. Ausencia de síntomas acompañantes y de otros antecedentes personales de interés. Como antecedentes familiares destaca que la madre fue intervenida a los 14 años de edad por feomocromocitoma bilateral. El estudio genético materno ha descartado mutaciones relacionadas con el protooncogén RET y los genes MAX y TMEN127.
En la exploración física del paciente se observa hipertensión arterial de 175/131mmHg (>P99/>P99). Se realiza también fondo de ojo, en el que se observa edema macular bilateral (OD>OI) con imagen de «estrella macular» por exudados duros.
Se lleva a cabo un extenso diagnóstico diferencial que incluye todos los tipos de HTA, tanto esencial como secundaria. El resto de la exploración física fue anodina. A su vez, las pruebas de laboratorio que incluyen hemograma, bioquímica y estudio de la función renal fueron normales.
Tras conocer el antecedente familiar materno, debemos descartar la presencia de un feocromocitoma. Se realiza ecografía-doppler abdominal donde se aprecia una masa de aproximadamente 4cm en el área renal hiliar izquierda, que depende de glándulas suprarrenales. Para completar el diagnóstico por imagen, se realiza tomografía axial computarizada (TAC) de abdomen donde se advierte una tumoración de 44×27mm anteroposterior con densidad heterogénea en su interior, que se desplaza desde fosa adrenal en sentido caudal, ventralmente a hilio vascular renal. También se realiza radiografía de tórax y se descarta extensión del tumor.
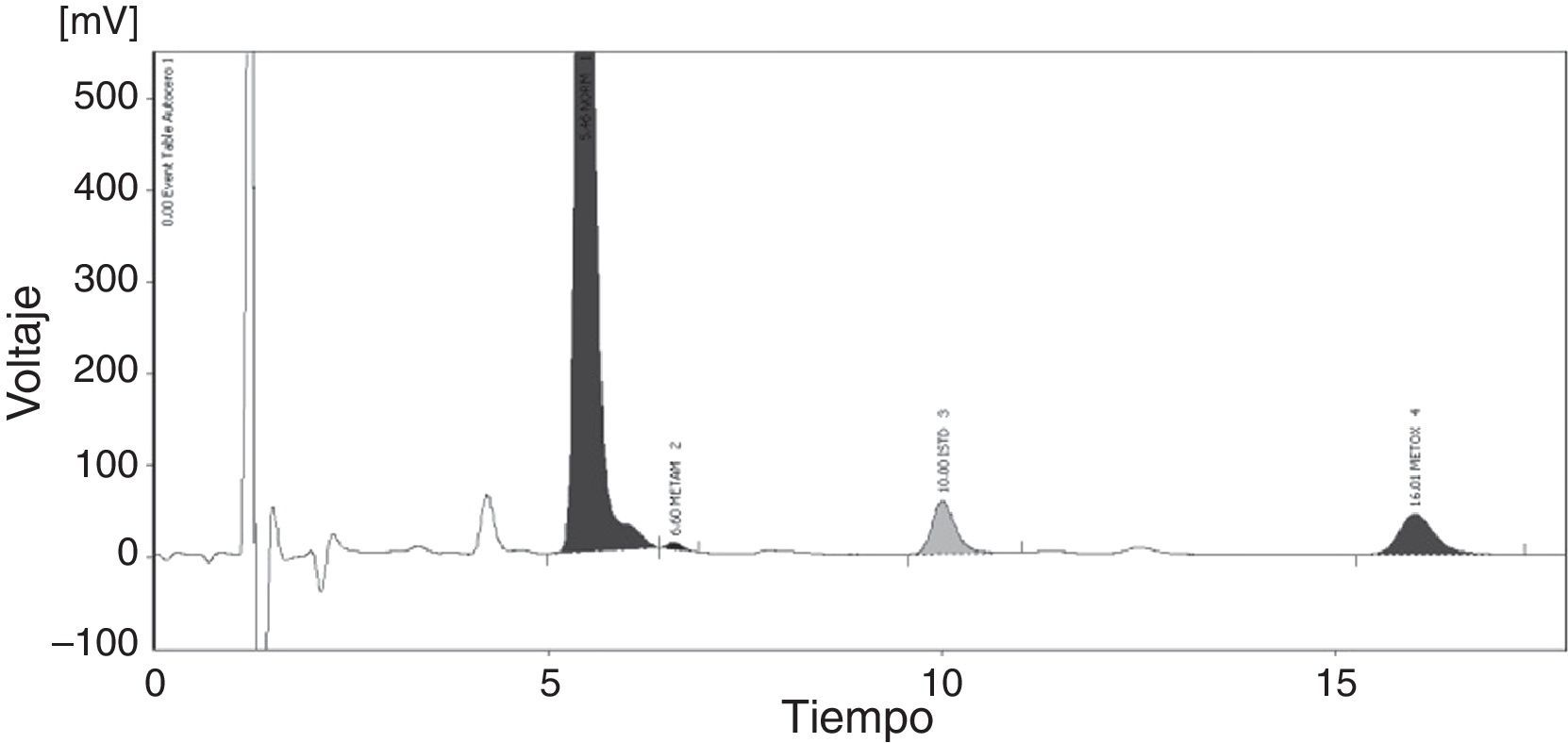
Simultáneamente, se solicita al laboratorio la determinación cuantitativa en orina de aminas biogénicas mediante cromatografía líquida de alta resolución (HPLC) (fig. 1). Los resultados se describen en la tabla 2, donde destacan niveles muy elevados de normetanefrinas y un moderado incremento de ácido vanilmandélico.

Teniendo en cuenta los datos de secreción bioquímica (tumor secretor de noradrenalina), la localización adrenal del tumor, la temprana edad al diagnóstico y que mutaciones específicas en el gen VHL pueden asociarse a feocromocitoma como única manifestación de la enfermedad, se decide comenzar el análisis molecular con el estudio de este gen, ligado a la enfermedad de VHL5. Se detecta una mutación en el exón 2 del gen VHL que genera un cambio de aminoácido (p.Phe119Leu), alterando la estructura cuaternaria de la proteína. Este cambio aminoacídico ha sido descrito previamente en la bibliografía consultada asociado a enfermedad de VHL tipo 26,7, sin embargo, no podemos asegurar que se comporte como VHL 2A, 2B o 2C: solo la evolución del paciente nos aportará esta información. Una vez identificada la mutación responsable de la enfermedad, es conveniente descartarla en otros miembros de la familia. Se realiza estudio genético en los familiares directos, y se identifica la misma mutación en la madre y en la hermana del caso índice, afectas de la enfermedad, por lo que se hace necesario un seguimiento estrecho por un equipo multidisciplinar.
En espera de los resultados genéticos y con el diagnóstico de masa suprarrenal indicativa de feocromocitoma, se decide su extirpación quirúrgica. Se inicia tratamiento preoperatorio con doxazosina (1,5mg/24h) y labetalol (50mg/12h) y a los 14 días se realiza con éxito la exéresis del tumor mediante laparoscopia, conservando la glándula suprarrenal. Durante el ingreso no presenta incidencias relevantes. A los 10 días el paciente es dado de alta con TA 96/53mmHg y se recomienda control y seguimiento ambulatorio. La anatomía patológica confirmó el diagnóstico de sospecha de feocromocitoma benigno.
DiscusiónEl diagnóstico de enfermedad de VHL debería sospecharse en aquellos casos con múltiples manifestaciones viscerales de la enfermedad, en pacientes con hemangioblastomas de la retina o del SNC, feocromocitomas de aparición precoz y en pacientes con familiares de primer grado diagnosticados de VHL4. Es necesario un seguimiento estrecho de estos pacientes y sus familias, con un protocolo de detección sistemático desde el primer año de vida.
Alrededor de un 20% de los pacientes con enfermedad de VHL desarrollan feocromocitomas (adrenales y extraadrenales). Aunque la incidencia de feocromocitomas es baja (1:100.000 pacientes/año), se describe un 20% en población pediátrica: se trata del tumor endocrino más frecuente de la infancia4,8. En las series pediátricas hay una clara diferencia respecto a los adultos en cuanto a presentación clínica: la HTA se encuentra en un 60-90% de los casos, normalmente mantenida y no asociada a episodios de crisis hipertensivas4,9,10.
La sospecha clínica de feocromocitoma debe seguirse del diagnóstico bioquímico, con determinación de catecolaminas y sus metabolitos (metanefrina, normetanefrina y ácido vanilmandélico) tanto en plasma como en orina de 24 h. Las metanefrinas/normetanefrinas son menos propensas a variaciones debidas a la actividad física del paciente, a la dieta, a la función renal o a la medicación. Su producción es continua en el tumor y son las que mejor reflejan la masa tumoral secretora. No obstante, las últimas recomendaciones indican que la cuantificación de metanefrinas libres en plasma y fraccionadas en orina mediante HPLC es el método diagnóstico de elección4,10. La localización inicial del tumor se realiza mediante técnicas de imagen (TAC o resonancia magnética). En caso de duda, se emplea la gammagrafía con meta-yodo-bencilguanidina con gran especificidad4. El tratamiento de elección es la resección del tumor mediante cirugía por vía laparoscópica, pero siempre después de una preparación farmacológica. El pronóstico es bueno, excepto en los casos de enfermedad maligna. Se recomienda seguimiento hormonal al mes de la cirugía, y luego anual de forma indefinida10.
Ante un paciente diagnosticado de feocromocitoma, debe considerarse su estudio genético, especialmente en los casos de feocromocitoma bilateral o extraadrenal, con edad menor de 45 años al diagnóstico o con historia familiar previa, porque este tipo de tumores suelen asociarse a síndromes genéticos familiares11.
Se aconseja iniciar el estudio de los feocromocitomas de localización adrenal en el gen VHL, seguido del protooncogén RET, dada la frecuencia de mutaciones descritas12. Tras el diagnóstico de enfermedad de VHL, es fundamental el estudio molecular a los familiares de primer grado. Se estima que aproximadamente el 80% de los casos con enfermedad de VHL tienen un progenitor afecto y que un 20% se debe a mutaciones de novo4,13. Por todo lo descrito anteriormente, es esencial realizar consejo genético a los pacientes y sus familiares una vez identificada la mutación.
Enlaces de interésThe VHL mutations database. Disponible en: http://www.umd.be/VHL
Alianza española de familias de VHL. Disponible en: http://www.vhl.org
FinanciaciónEl presente estudio no ha recibido financiación de ningún tipo, pública o privada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Centro Nacional de Investigaciones Oncológicas (Madrid) por los estudios genéticos realizados a la familia.
