



El aseguramiento de la calidad de la fase preanalítica se orienta hacia 2 aspectos clave: la gestión de los errores preanalíticos desde la perspectiva de la seguridad del paciente, y la mejora y armonización de los procedimientos, basada en la aplicación de normativa además de recomendaciones profesionales. Al igual que el resto de las fases, debe incluir un programa interno de aseguramiento y la participación en programas de intercomparación entre laboratorios.
El control de calidad interno debe basarse fundamentalmente en la identificación de riesgos, detección sistemática de errores y establecimiento de indicadores. La selección de los indicadores priorizando el impacto en el paciente, la forma de detectar y registrar los errores de forma sistemática y fácilmente explotable, así como las variables utilizadas en su cálculo, son aspectos importantes para medir la eficacia de las acciones de mejora y permitir la comparabilidad entre laboratorios. En este sentido, los programas externos de la calidad de la fase preanalítica basados en la comparación de indicadores, son una herramienta útil para el diseño e implantación de un programa de aseguramiento de la calidad.
Este documento pretende servir de apoyo para que cada laboratorio seleccione, implante y evalúe sus propios indicadores, de acuerdo a las características individuales de sus procedimientos preanalíticos, pero sin perder de vista la armonización entre laboratorios.
The quality assurance of the pre-analytical phase is oriented towards two key aspects; the management of pre-analytical errors from the perspective of patient safety, and the improvement and harmonisation of procedures, based on the application of regulations and professional recommendations. Like the rest of the phases, it should include an internal quality assurance program, as well as the participation in external quality assurance programs.
The internal quality control should mainly be based on the identification of risks, systematic detection of errors, and establishment of indicators. The selection of indicators prioritising the impact on the patient, the way to detect and record errors in a systematic and easily exploitable manner, and also the variables used in the calculations, are important aspects to measure the effectiveness of improvement actions and to allow comparability between laboratories. In this sense, the external quality assurance programs of the pre-analytical phase based on the comparison of indicators are a useful tool for the design and implementation of a quality assurance program.
This document is intended as a support for each laboratory to select, implement, and evaluate its own indicators, according to the individual characteristics of its pre-analytical procedures, but without losing sight of the harmonisation between laboratories.
El objeto de este documento es establecer recomendaciones prácticas para el diseño e implementación de un programa de aseguramiento de la calidad de la fase preanalítica. Estas recomendaciones se basan fundamentalmente en la detección, registro y control de los errores, priorizando la minimización de aquellos que puedan implicar un mayor impacto sobre la seguridad del paciente.
La mayoría de los errores producidos en el laboratorio clínico suceden en la fase extraanalítica, siendo la fase preanalítica la que concentra el mayor porcentaje de ellos, entre un 40-70% según diferentes autores. La gestión de la calidad de esta fase constituye, por tanto, una gran oportunidad de mejora para el laboratorio clínico1–4.
En las últimas décadas se ha producido una importante mejora de la calidad de la fase analítica gracias a la instauración sistemática de programas de control de calidad, y la implicación de los profesionales del laboratorio junto con la industria del diagnóstico in vitro en la mejora de reactivos y tecnología5. Los avances en la gestión de la calidad de la fase preanalítica se enfrentan a múltiples dificultades, ya que muchos de los procesos preanalíticos se desarrollan fuera del propio laboratorio, con personal muy numeroso y no dependiente del mismo, con una menor estandarización y automatización de los procesos, indicadores y especificaciones de la calidad menos consensuados y programas de evaluación externa limitados6.
Los avances en el aseguramiento de la calidad de la fase preanalítica se han dirigido en los últimos años hacia 2 aspectos clave: la seguridad del paciente y la mejora y armonización de procedimientos, apoyados en normativas específicas de laboratorio, como la Norma UNE-EN 151897, y en recomendaciones de los organismos científicos.
Desde que en 1999 el Institute of Medicine publicara el informe To err is human8, la seguridad del paciente ha sido uno de los objetivos estratégicos en la mejora de los sistemas sanitarios, siendo adoptada por organizaciones sanitarias tanto nacionales como internacionales.
Esta corriente ha llevado a la creación de nuevos modelos de gestión de la calidad orientados a la detección de los riesgos y la disminución de los errores. Se han desarrollado normas internacionales de aplicación concreta para la evaluación de riesgos9,10, incluyendo normativa específica para la gestión de riesgos orientada a la seguridad del paciente11. En el laboratorio de medicina se han incorporado estas estrategias a la práctica asistencial con especial repercusión en la fase preanalítica. En el ámbito nacional, la Comisión de Seguridad del paciente de la Sociedad Española de Medicina de Laboratorio (SEQCML) ha difundido diversos documentos12,13, incluyendo aspectos concretos de especial interés, como es la identificación segura de las muestras14.
En la seguridad del paciente, un aspecto fundamental es valorar el impacto de los errores de laboratorio en el paciente. Snydman et al.15 realizaron en 2012 un análisis sobre 266.224 errores producidos entre los años 2000-2005 en 30 organizaciones sanitarias de EE. UU. Basándose en un sistema de registro de incidentes de seguridad global de estas instituciones encuentran que los errores de laboratorio representaron uno de cada 7 eventos. Dentro de los incidentes de seguridad en el laboratorio la fase preanalítica es la principal fuente, con un 81% de los mismos. Respecto a su impacto en el paciente, el 45% de los errores llegaron al paciente, generando daño o incrementando el número de pruebas necesarias para su seguimiento. Cabe destacar que el 8% de los incidentes ocasionaron un daño temporal y que el 0,08% provocaron daño permanente o muerte15.
Respecto a la armonización de los procesos preanalíticos, una vía fundamental para demostrar competencia técnica y generar confianza en los usuarios del laboratorio es la aplicación de la norma UNE-EN ISO 15189:2013. En ella se indica que el laboratorio debe asegurar la calidad de los análisis implementando procesos adecuados y verificando su eficacia mediante el uso de indicadores apropiados que permitan la mejora continua. Todos los procesos, incluidos los preanalíticos, deberán formar parte del sistema de gestión de la calidad. Asimismo, recomienda la participación en programas de comparación entre laboratorios que verifiquen el proceso completo. La Comisión de Acreditación de Laboratorios y la de Calidad Extraanalítica de la SEQCML han publicado conjuntamente un documento de recomendaciones para la gestión de los procesos preanalíticos según la Norma International Organization for Standardization (ISO) 15189:2013, donde se contemplan los requisitos de dicha norma y los aspectos prácticos de su implantación en el laboratorio clínico16.
Otro aspecto importante en la armonización es la participación en programas de evaluación externa de la calidad. La participación en estos programas sirve de guía y ofrece la posibilidad de evaluar la mejora de la calidad preanalítica. Esta evaluación comparativa se incluye en los estándares de certificación-acreditación de diferentes agencias y organismos (College of American Pathologists, International Organization for Standardization, Joint Comission, etc.). Desde hace 20 años la SEQCML organiza uno de los pocos programas que existen de evaluación externa de la calidad de la fase preanalítica. Este programa tiene como objetivo la comparación de indicadores de la calidad de la fase preanalítica y permite establecer especificaciones de la calidad basadas en el estado del arte17.
A nivel internacional diversos grupos, como el Working Group for preanalytical phase de la European Federation of Clinical Chemistry and Laboratory Medicine (EFLM), o el Working Group Laboratory errors and patient safety (WG-LEPS) de la International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) están involucrados activamente en proyectos de mejora y armonización de la fase preanalítica, destacando entre otros la estandarización de procedimientos como la obtención de muestras, identificación del paciente o la armonización de los indicadores de la calidad18–22.
Control de calidad internoErrores preanalíticosSe define error preanalítico como cualquier evento que ocurre antes del análisis de la muestra y que puede comprometer la exactitud del resultado y/o la seguridad del paciente.
Identificación de errores y puntos críticos. Mapa de riesgosCada laboratorio debe determinar los puntos críticos de sus procesos y establecer su mapa de riesgos7. La identificación de puntos críticos y la evaluación de riesgos puede llevarse a cabo mediante el uso de herramientas de análisis de seguridad del paciente. Podemos distinguir 2 tipos de herramientas, las de tipo reactivo y las de tipo proactivo. Entre las primeras están el registro de fallos y sistema de acciones correctivas, o el análisis de causas raíz, ambas basadas en reducir la frecuencia de errores ya una vez originados. Entre las segundas destacan el análisis modal de fallos y efectos (AMFE) que se basa en la reducción del riesgo de fallos potenciales23–25.
La metodología AMFE se ha aceptado como método de elección para la evaluación de riesgos en el documento ISO/TS 2236710, así como en la norma UNE-EN ISO 15189:2013. El College of American Pathologists ha desarrollado una guía para la evaluación de riesgos que puede ser de utilidad para su aplicación en el laboratorio26.
Los métodos proactivos tipo AMFE tienen como objetivo la prevención y priorización de los riesgos detectados. Esta metodología se recomienda por su mejor aceptación entre los profesionales, ya que, al incidir en el diseño del proceso, subproceso o tarea, facilita su desempeño y comunicación23. Las limitaciones de este método pueden compensarse mediante la utilización de alguna de las herramientas de tipo reactivo como el análisis de causas raíz o el registro de fallos y sistema de acciones correctivas que actúan sobre los errores detectados según su frecuencia y gravedad. Como en todo ciclo de mejora continua, las acciones de mejora o preventivas deben evaluarse periódicamente, modificando el mapa de riesgos original.
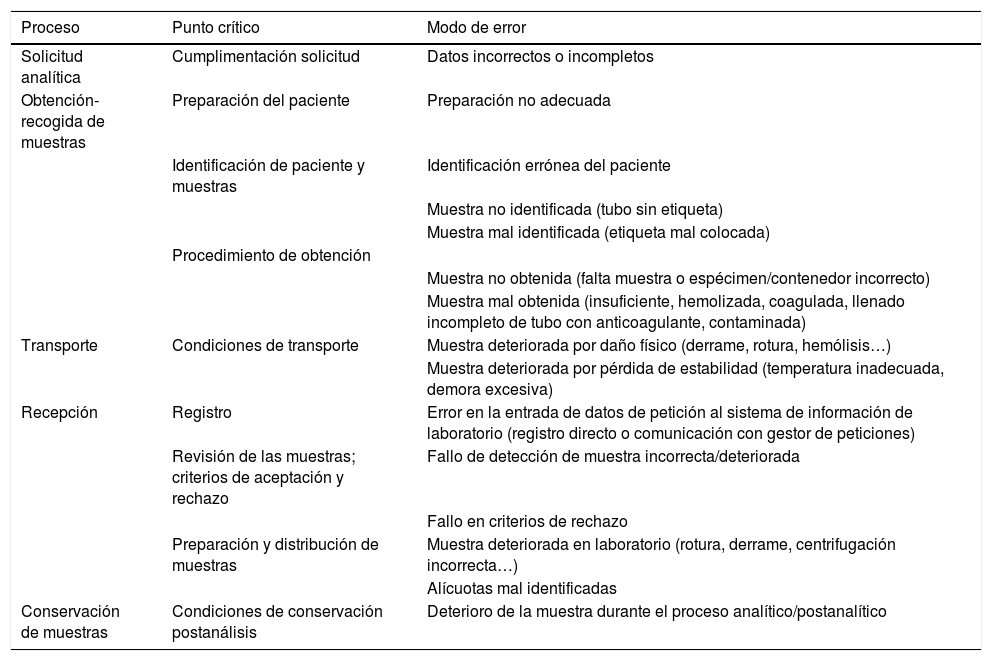
Cada laboratorio debe establecer su propio mapa de riesgos basado en la localización de sus puntos críticos y la priorización de los modos de fallo, asignando la frecuencia y repercusión a cada uno de ellos. Se presenta un ejemplo de esquema básico de clasificación de los procesos preanalíticos con identificación de los puntos críticos y tipos de error habituales en la tabla 1.
Ejemplo de clasificación de los procesos preanalíticos e identificación de puntos críticos y modos de error
| Proceso | Punto crítico | Modo de error |
|---|---|---|
| Solicitud analítica | Cumplimentación solicitud | Datos incorrectos o incompletos |
| Obtención-recogida de muestras | Preparación del paciente | Preparación no adecuada |
| Identificación de paciente y muestras | Identificación errónea del paciente | |
| Muestra no identificada (tubo sin etiqueta) | ||
| Muestra mal identificada (etiqueta mal colocada) | ||
| Procedimiento de obtención | ||
| Muestra no obtenida (falta muestra o espécimen/contenedor incorrecto) | ||
| Muestra mal obtenida (insuficiente, hemolizada, coagulada, llenado incompleto de tubo con anticoagulante, contaminada) | ||
| Transporte | Condiciones de transporte | Muestra deteriorada por daño físico (derrame, rotura, hemólisis…) |
| Muestra deteriorada por pérdida de estabilidad (temperatura inadecuada, demora excesiva) | ||
| Recepción | Registro | Error en la entrada de datos de petición al sistema de información de laboratorio (registro directo o comunicación con gestor de peticiones) |
| Revisión de las muestras; criterios de aceptación y rechazo | Fallo de detección de muestra incorrecta/deteriorada | |
| Fallo en criterios de rechazo | ||
| Preparación y distribución de muestras | Muestra deteriorada en laboratorio (rotura, derrame, centrifugación incorrecta…) | |
| Alícuotas mal identificadas | ||
| Conservación de muestras | Condiciones de conservación postanálisis | Deterioro de la muestra durante el proceso analítico/postanalítico |
La detección y notificación de errores es la base de un sistema de gestión orientado a la seguridad del paciente. Se debe asegurar que todos los errores se detectan y registran en un sistema que permita su estudio y explotación. El registro de los errores detectados a lo largo del proceso preanalítico resulta indispensable para la obtención de la información necesaria en el cálculo de los indicadores, que como veremos son la base del aseguramiento de la calidad en la fase preanalítica. Debido a los múltiples procesos y personal involucrado, las variables a controlar son numerosas y difíciles de definir, por lo que es necesario que el laboratorio disponga de un procedimiento sistematizado de registro y comunicación de errores.
Es recomendable que este registro esté integrado en el sistema informático del laboratorio (SIL). De esta forma se facilita que todo el personal participe en la detección y registro de errores en cualquier parte del proceso total del laboratorio para asegurar la máxima detección posible. Es aconsejable que los distintos errores o incidencias se estructuren por medio de códigos asociados a textos predefinidos que describan perfectamente y con precisión el error ocasionado, facilitando la remisión de la información al origen y su posterior estudio y resolución.
La detección de los errores podrá ser humana, ya sea durante la realización de los procesos o en las fases de verificación, o utilizando sistemas preanalíticos automatizados que pueden detectar muestras deterioradas o incongruencias entre las muestras y las solicitudes. Los sistemas automatizados pueden además enviar esta información directamente al SIL, dando lugar a un procedimiento de registro más sistemático que mejore la tasa de detección. En el registro manual de errores, los datos, consignados en impresos de registro, deben ser introducidos en el SIL para su análisis, lo que limita y ralentiza su comunicación y evaluación. Por este motivo se recomienda la utilización de sistemas automatizados para la gestión de incidencias preanalíticas.
Indicadores de la calidad de la fase preanalíticaUn indicador se define como «Datos o conjunto de datos que ayudan a medir objetivamente la evolución de un proceso o de una actividad» (Norma UNE 66175: 2003)27.
La Norma UNE-EN ISO 15189:2013, en su apartado 4.14.7 «Indicadores de la calidad», refiere: «el laboratorio debe establecer indicadores de la calidad para realizar el seguimiento y evaluar el desempeño, observando los aspectos críticos de los procesos preanalíticos, analíticos y postanalíticos».
Los indicadores son herramientas de gestión de procesos, que permiten conocer el estado y las capacidades de mejora de los mismos a través de su seguimiento y análisis. Estos indicadores deben estar relacionados con los procesos, aportando información sobre su tendencia y sobre el cumplimiento de las especificaciones previstas. De esta forma, pueden aplicarse los reajustes necesarios que permiten una mejora continua. Los indicadores son, por tanto, una herramienta clave para medir la calidad y comprobar si se han conseguido los objetivos definidos.
Características de los indicadoresLas características de un indicador pueden resumirse con el acrónimo SMART (eSpecífico, Medible, Alcanzable, Realista y en Tiempo). Según la Norma UNE 66175:2003 los indicadores deben reunir una serie de características:
- •
Que representen una actividad importante o crítica (puntos clave).
- •
Fiables.
- •
Alcanzables.
- •
Con resultados cuantificables y transferibles al valor del objetivo.
- •
Estables y válidos en el tiempo, para ver su evolución.
- •
Intercomparables con otras organizaciones (indicadores consensuados).
- •
Que compense el esfuerzo (buena relación coste/beneficio).
Aunque existen propuestas de indicadores estandarizados, que se revisarán posteriormente, es recomendable que cada laboratorio seleccione y priorice los indicadores a utilizar dependiendo de su definición de proceso preanalítico y su mapa de riesgos.
La forma de cálculo y la frecuencia de evaluación del indicador dependerá del uso previsto y del riesgo asociado al proceso controlado, pudiendo distinguir varios tipos28:
- •
Centinela: vigilancia de errores de gran gravedad para el paciente.
- •
De puntos críticos: en relación con el mapa de riesgos establecido por el laboratorio.
- •
De eficacia: evalúan el grado de calidad de los resultados del proceso.
- •
De eficiencia: comparan resultados respecto a recursos.
Es conveniente calcular los indicadores de forma relativa a la actividad, en forma de frecuencia o tasa. Se debe seleccionar un numerador que cuantifique la incidencia que se desea medir y un denominador que refleje el conjunto de actividades sometidas a riesgo. Para que los indicadores sean comparables entre diferentes laboratorios deben consensuarse tanto numerador como denominador. La definición del numerador debe ser clara y referirse a un modo de fallo concreto. La elección del denominador es también importante. Debe reflejar la misma actividad que el numerador y ser fácilmente medible y comparable entre laboratorios. La mayoría de los indicadores preanalíticos pueden expresarse respecto al número de pacientes, muestras o pruebas. La selección de distinto denominador puede dificultar mucho la comparación y establecimiento de especificaciones comunes.
Para obtener una información más específica, los indicadores deberían poder ser desagregados según el tipo de actividad (programada/urgente, hospitalización/ambulatoria), reflejando las diferencias entre distintos subprocesos y actores involucrados.
El laboratorio debe decidir y priorizar, en función de sus recursos, el número, tipo y frecuencia de indicadores preanalíticos. Un excesivo número de indicadores puede suponer una sobrecarga que limite su utilización.
Como veremos posteriormente, los indicadores son también la base de los programas de intercomparación entre laboratorios. Este es un criterio muy importante a la hora de decidir los indicadores a utilizar. Utilizar indicadores interoperables asegura su fiabilidad al contar con validación previa y facilita su evaluación.
A continuación se presentan los indicadores más habituales en la fase preanalítica como orientación para su selección.
a) Indicadores relacionados con errores en la identificación:
Este tipo de errores se consideran centinela por el impacto que pueden tener sobre la seguridad del paciente, aunque pueden incluir situaciones de distinto riesgo. Es interesante medir este indicador desglosando el número de errores de identificación detectados antes y después de la emisión de resultados, informando de la capacidad de detección del error en el laboratorio.
Peticiones mal identificadas. Se refiere a la discordancia entre el registro electrónico y la petición en formato papel. Este error se producía habitualmente en el registro manual de volantes en el laboratorio, y con la introducción de la petición electrónica ha disminuido notablemente. No obstante, siguen ocasionándose errores en los casos en los que es necesaria la conciliación del número de petición con el de identificación del laboratorio (etiqueta con código de barras que relaciona la solicitud con las muestras). Esta conciliación suele realizarse en los centros de extracción, fuera del laboratorio, por lo que el control del proceso se complica. Algunos sistemas asignan y asocian automáticamente el número de laboratorio con el código de la solicitud en el momento de la obtención de muestras, con lo que se minimiza este tipo de error.
Muestras mal identificadas. Este indicador hace referencia a las muestras que presentan discrepancias con la identificación del paciente o de la petición. Supone la asignación de los resultados de un paciente a otro distinto. Se trata por tanto de un indicador centinela y debe ser tratado con tolerancia cero.
Este error puede ser detectado durante la fase de validación por la no concordancia de datos demográficos y resultados (PSA elevado en mujeres, por ejemplo) o por discrepancia con resultados anteriores.
Muestras mal etiquetadas. Son muestras sin etiqueta de identificación, con etiqueta incorrecta respecto al tipo de código de barras (códigos de barra con sufijos o prefijos específicos según el tipo de muestra), o con etiqueta dañada haciéndola ilegible. Son incidencias de identificación de muestras que impiden la realización de pruebas, y aunque realmente no provocan un riesgo grave en el paciente, como sucede en el caso de la identificación errónea, suelen obligar a la repetición de la extracción.
b) Indicadores relacionados con la incorrecta cumplimentación de la solicitud y los errores de transcripción:
Se refiere a la falta de información necesaria para la realización o interpretación del análisis, como es la talla, peso o la diuresis. También puede monitorizar la falta de datos del solicitante (médico, servicio, etc.). Estos errores están asociados a las peticiones de tipo manual. También pueden hacer alusión al error en el registro de las pruebas solicitadas.
Los errores producidos por una transcripción manual pueden originarse en el laboratorio o fuera del mismo, por lo que puede ser interesante medirlos separadamente.
c) Indicadores relacionados con la calidad de la muestra:
Muestras inadecuadas y/o deterioradas. Estos indicadores están relacionados con la obtención de la muestra. Calculado de forma global puede ser utilizado para observar la tendencia general en el tiempo y el impacto de las intervenciones realizadas, como es el cambio de dispositivos o procedimiento en la obtención de muestras. Desagregado según el origen de las muestras, permite la comparación entre centros o personas implicadas y evalúa las acciones concretas como son la educación o el entrenamiento del personal extractor.
Cada laboratorio debe definir los criterios de aceptación y rechazo, que idealmente se deberían aplicar en la fase de revisión, antes de procesar las muestras, pero que muchas veces se detectan durante la fase analítica. Cada laboratorio debe además definir el grado de deterioro que supone el motivo de rechazo (grado de hemólisis, nivel de llenado del tubo de coagulación…). Esto puede suponer que ante un mismo problema un laboratorio opte por rechazar la muestra, lo que implica que una o varias determinaciones no pueden ser informadas, mientras que otro laboratorio puede optar por informar el resultado incluyendo o no una nota en el informe que indique una posible limitación. Es importante tener en cuenta la variabilidad en estos criterios a la hora de comparar los resultados entre laboratorios.
Es aconsejable definir de forma separada indicadores para cada tipo de muestra y motivo de deterioro. Los tipos de muestra utilizados en el laboratorio suelen tener formas habituales de deterioro (hemólisis para las muestras de suero o plasma, coágulos para las muestras de sangre completa, incorrecto volumen de llenado para la coagulación...). Un aspecto importante es la selección del denominador en estos casos. Al separar por tipo de muestra sería ideal compararlos con el número total de tubos de cada tipo, pero este es un dato que puede ser difícil de obtener y que puede variar entre laboratorios (algunos laboratorios pueden obtener varios tubos del mismo tipo en cada extracción). En el Programa de Garantía Externa de la fase preanalítica de la SEQCML se ha optado por referir estos errores en la muestra al número de pruebas solicitadas más representativas de cada espécimen. Por ejemplo, creatinina para suero, hemograma para el tubo con EDTA o tiempo de protrombina para el tubo con citrato. Estos datos son fáciles de obtener y mejoran la comparación de los indicadores entre laboratorios.
Las causas de deterioro de mayor importancia y frecuencia son:
- •
Muestra hemolizada: es el principal motivo de rechazo de muestras de suero. Como hemos comentado, cada laboratorio debe definir cuál es el grado de hemólisis que supone motivo de rechazo para una magnitud concreta. La automatización de la medida de los índices séricos en los sistemas analíticos ha mejorado la detección y la cuantificación de la hemólisis. No obstante, la falta de estandarización de la medida de los índices séricos en los distintos equipos analíticos, así como la expresión de resultados, sigue representando un problema29. Actualmente existen programas de intercomparación basados en material de control para evaluar las prestaciones de los índices séricos, como el organizado por la SEQCML. La utilización de índices séricos de forma sistemática ha permitido además generar un indicador global de calidad de la muestra basado en la detección de hemólisis de bajo grado, que sin significar motivo de rechazo indica una obtención o procesamiento inadecuado de la muestra. El indicador de índice hemolítico recomendado para este fin es el porcentaje de muestras de suero con IH≥0,5g/l.
- •
Muestra coagulada: debido habitualmente a un mal procedimiento en la obtención, extracción dificultosa o al no mezclar adecuadamente la sangre con el anticoagulante tras la extracción.
- •
Contenedor incorrecto, muestra inadecuada: si el contenedor utilizado no es correcto, la muestra puede resultar incompatible con las pruebas solicitadas, por ejemplo falta de intercambiabilidad entre suero y plasma o jeringa para gasometría mal cerrada.
- •
Muestra contaminada: debido a contaminación con líquidos presentes en la vía de extracción o con el aditivo de otros tubos. El primer supuesto es frecuente en el paciente hospitalizado y el segundo tiene que ver con mezcla de muestra obtenida en distintos tubos, como por ejemplo la contaminación por EDTA en tubos de suero.
- •
Muestra inadecuada por incorrecta preparación: debido a una preparación incorrecta del paciente (ayuno, falta de preparación específica requerida, reposo, muestras de orina recogidas sin el conservante adecuado, etc.). Este error pone en evidencia la importancia de suministrar unas instrucciones adecuadas al paciente.
- •
Centrifugación incorrecta: presencia de restos de fibrina en tubos de suero o exceso de plaquetas en el tubo de coagulación. Se puede deber a fuerza centrífuga incorrecta o a coagulación incompleta en los tubos de suero.
- •
Conservación incorrecta, pérdida de estabilidad: muestra que ha sobrepasado el límite de estabilidad para una magnitud. El límite de estabilidad debe establecerse de forma separada para el espécimen (sangre completa por ejemplo) y para la muestra (suero o plasma por ejemplo). El deterioro puede ocurrir durante el transporte o en el propio laboratorio, ya sea en la fase analítica o durante su conservación en seroteca para ampliación de pruebas. Es un indicador muy asociado al transporte de muestras, donde se detecta por los registros de tiempo y temperatura, pero que también puede ser detectado por resultados anómalos, como un descenso importante de glucosa acompañado de un exagerado aumento de fósforo en una muestra de sangre con más de 24horas sin centrifugar, o como elevación sistemática de potasio en muestras de sangre completa transportadas a baja temperatura.
d) Indicadores relacionados con el volumen de la muestra:
Muestra no recibida. Muy frecuente en las muestras que debe obtener el propio paciente, como es el caso de la orina. Puede diferenciarse en este caso la muestra no entregada voluntariamente por el paciente de la muestra no recibida por motivos achacables al centro de extracción o laboratorio.
Es muy importante contar con listados o sistemas informáticos de soporte a la extracción que indiquen al personal de toma de muestras el número y tipo de especímenes a obtener.
Muestra insuficiente. Muy frecuente en solicitudes muy amplias o en niños debido al intento de reducción de la cantidad de muestra obtenida para evitar sangría excesiva al paciente.
El laboratorio debe tener en cuenta el número total de tubos necesarios para solicitudes con pruebas de áreas diferentes, de forma que el volumen sea suficiente, pero evitando el exceso de volumen extraído. La recepción y preparación centralizada de las muestras permite gestionarlas de forma más eficiente, compartiendo tubos o alícuotas en su caso en diferentes áreas del laboratorio.
Volumen de muestra incorrecto respecto al anticoagulante. Principalmente referido a la muestra de coagulación incorrectamente enrasada.
e) Indicadores relacionados con el transporte:
Muestras rechazadas por incorrectas condiciones de transporte o conservación. Las causas principales son el tiempo y/o temperatura inadecuados. Otras causas que se pueden considerar son las roturas de contenedor o derrames asociados a la agitación durante el transporte. Es interesante diferenciar entre transporte externo (desde centros de extracción, habitualmente por carretera) e interno (a mano o mediante tubo neumático).
La detección de este tipo de incidencias requiere trazabilidad del momento de obtención y del tiempo de transporte unido a un sistema de registro de temperatura.
Como ya hemos comentado, cada laboratorio debería definir los límites de estabilidad para cada magnitud y, a partir de la cartera ofertada, calcular los límites generales para cada espécimen y establecer las magnitudes que requieren un tratamiento especial.
f) Indicadores de seguridad laboral relacionados con la fase preanalítica:
Número de accidentes laborales registrados. Requieren un tratamiento específico, muchas veces regulado por los servicios de prevención de riesgos, y supone un indicador centinela que debe hacer revisar las medidas de protección establecidas. Incluye el pinchazo accidental al realizar extracciones o la contaminación de mucosas por salpicadura.
g) Indicadores de resultado:
Los indicadores de resultado son aquellos relacionados con los resultados globales de salud. Por ejemplo, la mejora en el manejo de muestras de control ambiental de microbiología respecto a la disminución de las infecciones asociadas a la asistencia, o el tiempo de respuesta de las peticiones urgentes respecto al tiempo de estancia del paciente en urgencias, etc., NO son fáciles de obtener, y se recurre muchas veces a mediciones indirectas, como pueden ser el grado de satisfacción de usuarios y pacientes (mediante encuesta, por ejemplo), el número de reclamaciones presentadas por usuarios y pacientes o el tiempo de demora para la cita de extracción.
h) Indicadores globales del proceso preanalítico:
Número total de errores detectados durante el proceso preanalítico. Este indicador puede resultar útil para analizar la tendencia o evolución general de un laboratorio o para establecer comparaciones entre organizaciones.
Suele distinguirse entre: incidencias preanalíticas extralaboratorio, que es todo aquello relacionado con la cumplimentación de la solicitud, la identificación del paciente y las muestras, la preparación del paciente, la obtención y recogida de especímenes y su transporte y las incidencias intralaboratorio, relacionadas con el registro interno de las solicitudes y el procesado y conservación de las muestras hasta su análisis.
Planificación e implementaciónUna vez se han seleccionado los indicadores se deberá planificar su implementación en la organización, definiendo una serie de aspectos según las necesidades del laboratorio. A este respecto, la norma UNE-EN ISO 15189:2013 en su apartado 4.14.7 «Indicadores de la calidad» establece que: «se debe planificar el proceso de realizar el seguimiento de los indicadores de la calidad, lo que incluye establecer objetivos, metodología, interpretación, límites, plan de acción y duración de la medición».
La implementación debe involucrar y motivar a las personas afectadas, mediante la formación y comunicación de los resultados.
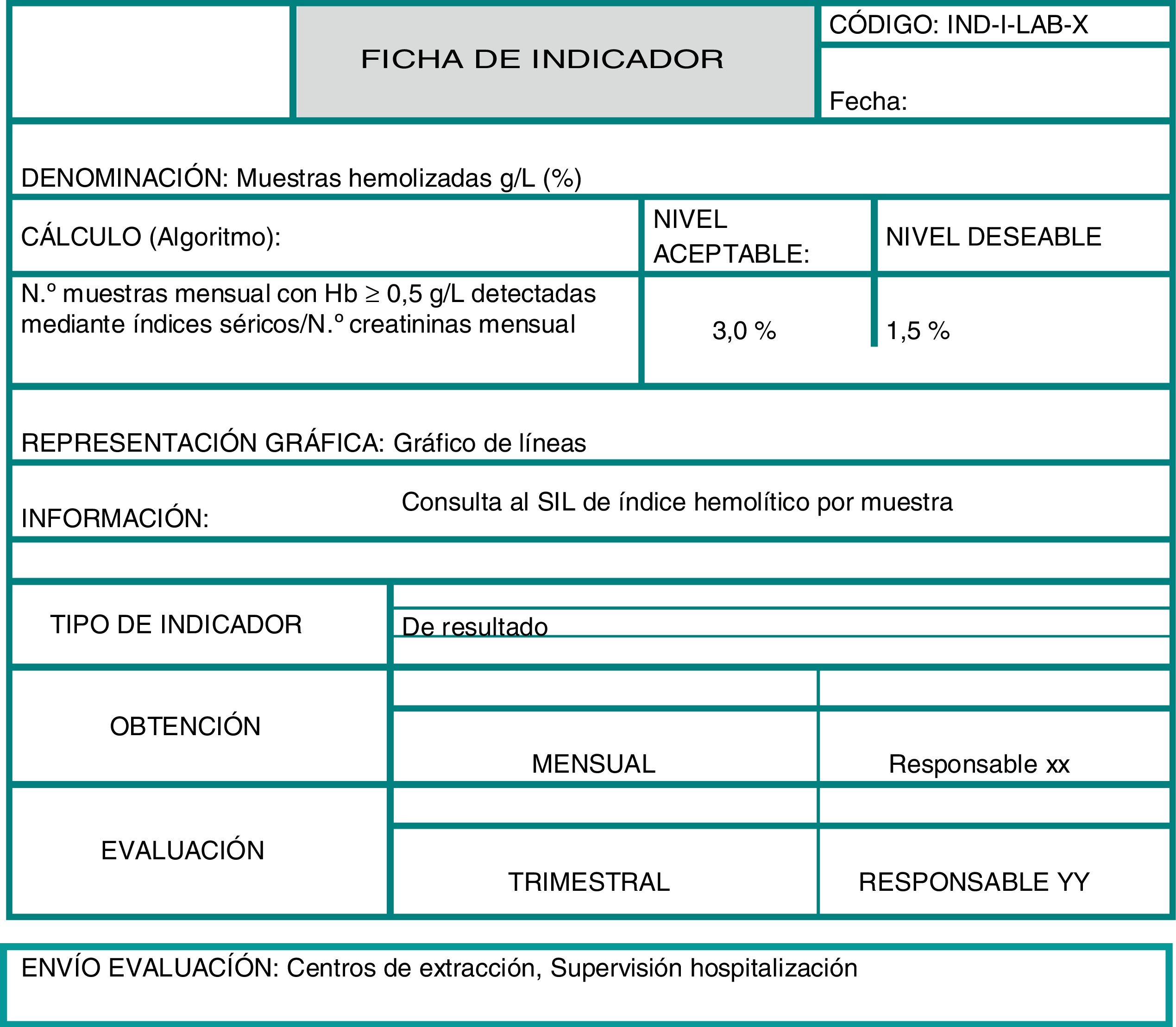
Para formalizar los indicadores resulta muy útil elaborar una ficha en la que se describan todos los aspectos mencionados y que se hayan considerado en cada caso. A continuación, en la figura 1, se muestra la información que puede contener una ficha de indicador a modo de ejemplo.
Validación y revisión
La validación del indicador se debe realizar tras la obtención de resultados, comprobando si se han cumplido los objetivos previstos y si el esfuerzo de obtención ha sido rentable. Asimismo, se debe consultar sobre su idoneidad a los usuarios. La evaluación periódica de los indicadores establecidos nos ofrece información sobre los resultados relevantes del indicador como, por ejemplo, su tendencia en el tiempo (positiva o negativa), ya que a pesar de que se cumplan las especificaciones previstas, nos informará del grado de mejora o deterioro respectivamente.
Esta evaluación se debe registrar para facilitar su seguimiento e informar a los interesados, recogiendo posibles causas de cambio, medidas correctivas o preventivas propuestas o personas a quien se debe comunicar.
Hay que tener en cuenta los posibles cambios organizativos del laboratorio o su propia evolución en el tiempo, ya que los indicadores pueden quedar obsoletos, bien porque no informan de aspectos útiles o relevantes, bien porque en definitiva no informan sobre los objetivos.
Especificaciones de la calidad para los indicadores preanalíticosLas especificaciones de la calidad pueden definirse como: «el valor de una medida que no se quiere sobrepasar, en función de un requisito preestablecido», o como «el nivel de desempeño requerido para facilitar la toma de decisiones clínicas»30.
El laboratorio debe establecer las especificaciones de la calidad o límites de aceptabilidad para cada uno de los indicadores y establecer las acciones correctivas derivadas de su incumplimiento.
La jerarquía de modelos para establecer las especificaciones de la calidad analítica fue definida en la Conferencia de Estocolmo en el año 1999. Más tarde, en el año 2014 en la Conferencia de Milán, se revisó el esquema jerárquico, pasando de 5 a 3 modelos. A partir de esta conferencia se creó el grupo de trabajo Finish Group on Performance Specifications for the extra-analytical phases (TFG-PSEP) de la EFLM con la intención de definir el modelo adecuado para establecer las especificaciones de la calidad de las fases extraanalíticas31.
Siguiendo el esquema jerárquico establecido en Milán, las especificaciones podrán definirse de forma escalonada por: 1) el efecto sobre los resultados clínicos; 2) la variabilidad biológica; y 3) el estado del arte. Es este último criterio el indicado por el TFG-PSEP para la selección de especificaciones de la fase preanalítica.
La selección de especificaciones según el estado del arte puede basarse en datos del desempeño de otros laboratorios de características semejantes reportados en la literatura, pero es preferible basarse en los resultados de programas de evaluación externa de la calidad de la fase preanalítica.
El trabajo del grupo de laboratorios del Institut Català de la Salut, publicado en 2011, ofreció resultados de sus indicadores de la calidad para procesos clave analíticos y extraanalíticos entre los años 2004 y 2008, así como una propuesta de especificaciones de la calidad basadas en los resultados obtenidos en una primera fase. Posteriormente se transformaron los valores a una escala Seis Sigma para detectar los procesos que requerían mejora. A partir de los cálculos efectuados se consideraron procesos bien controlados aquellos en que sigma resultó≥432.
Una forma habitual de presentar los resultados de los programas de intercomparación es el uso de percentiles para expresar el posicionamiento de un laboratorio respecto al grupo de comparación. A iniciativa del TGF-PSEP se ha establecido de forma consensuada entre la IFCC y la EFLM un criterio para establecer las especificaciones de la calidad preanalíticas basadas en los resultados de los laboratorios participantes en el programa de indicadores de la calidad de la IFCC. Exponen que, para un periodo anual, se pueden calcular los percentiles 25, 50 y 75 de toda la distribución de laboratorios participantes en el programa y establecer de esta forma 3 niveles de desempeño en relación con el grado de calidad:
- -
Resultados inferiores al percentil 25: alto grado de calidad o desempeño (óptimo).
- -
Resultados entre los percentiles 25 y 75: grado de calidad o desempeño medio (deseable).
- -
Resultados superiores al percentil 75: baja calidad o desempeño (mínimo).
Se debe tener en cuenta que, si el indicador elegido determina la incidencia de errores, un resultado elevado del indicador nos informará sobre una peor calidad. Por el contrario, si el indicador está relacionado con la medida de la mejora o situación satisfactoria, la escala se invertiría, relacionando un mayor percentil con una mayor calidad.
Se ha establecido que los resultados de cada año serán utilizados como especificaciones durante el siguiente, salvo que no mejoren respecto a los previos, en cuyo caso no se modificarán33.
El programa de garantía externa de la calidad de la fase preanalítica de la SEQCML presenta los resultados de la evaluación del grupo de laboratorios participantes de forma similar, incluyendo los percentiles 10, 25, 50, 75 y 9034.
A pesar de la conveniencia de disponer de especificaciones para los indicadores de la calidad preanalítica, es importante considerar que la mayoría de los errores preanalíticos son evitables. Por lo tanto, un objetivo ideal sería la erradicación total de los mismos. Algunos errores, como es el caso de la identificación inequívoca del paciente, ya se ha definido previamente que deben tratarse como indicadores centinela y debe perseguirse el objetivo de «error cero», y no solo el de aceptar el estado del arte.
Armonización de indicadoresEn el año 2008 el grupo WG-LEPS de la IFCC comenzó un proyecto de definición de un modelo común de indicadores de la calidad, con un método armonizado de recogida de datos, gestionado como un programa de garantía externa de la calidad, e incluyendo todas las fases del proceso analítico. Estos indicadores debían cumplir con los requerimientos de la norma UNE-EN ISO 15189:2013 y ser representativos de todas las actividades críticas, además de ser medibles por una mayoría de laboratorios a nivel mundial independientemente del tipo de laboratorio, número y tipo de pacientes, tipo de actividad, formación del personal, etc. La primera fase del proyecto se desarrolló entre los años 2008 y 2013 mediante la participación libre y anónima de los laboratorios a través de una Web de la IFCC, sin necesidad de participar en la totalidad de los indicadores propuestos3.
En la conferencia de Padua del año 2013 se realizó una revisión de la experiencia de 5 años, considerando la idoneidad o no de los indicadores y el procedimiento de recogida de datos. Se consensuó la asignación de prioridades de dichos indicadores para orientar a los laboratorios en su incorporación gradual en la práctica, refiriéndolos como obligados (prioridad 1), importantes (prioridad 2), sugeridos (prioridad 3) y valorables (prioridad 4). Los indicadores con prioridad alta evalúan etapas fundamentales y deben ser medidos por todos los laboratorios, independientemente de su tamaño y área geográfica que observan.
Se evaluaron los datos de 59 laboratorios que enviaron resultados durante los años 2014, 2015 y primer semestre de 2016, estableciéndose para cada uno de los 3 períodos los niveles de desempeño comentados en el punto anterior. Además, para expresar el nivel de calidad del proceso se calculó el nivel de calidad sigma35.
En 2016, en un esfuerzo conjunto entre el TFG-PSEP y el WG-LEPS, tuvo lugar una nueva conferencia consenso con participantes de 14 países, incluido España, IFCC, EFLM, fabricantes de diagnóstico in vitro, etc., en la que se alcanzó un consenso sobre los indicadores y especificaciones a utilizar en los laboratorios, sobre la base de cumplir requisititos de UNE-EN ISO 15189:2013, monitorizar las actividades críticas y promover la minimización de los riesgos de error. En la tabla 2 se detallan los indicadores seleccionados para 2017 y para la fase preanalítica, con nivel de prioridad de 1 y 2, expresados todos ellos en porcentaje. Los términos del acuerdo, así como el listado completo de indicadores por prioridades, pueden encontrarse en la Web del WG-LEPS, cuyo enlace se incluye en la bibliografía33.
Indicadores de la fase preanalítica con prioridad 1 y 2 consensuados en la Conferencia de Padua de 2016
| Indicador | Fórmula de cálculo | Prioridad |
|---|---|---|
| Errores de identificación de paciente | N.° de peticiones mal identificadas/n.° total de peticiones | 1 |
| N.° de muestras mal identificadas/n.° total de muestras | 1 | |
| Peticiones con pruebas inapropiadas | N.° de peticiones sin motivo clínico/n.° total de peticiones | 2 |
| Errores de transcripción de pruebas | N.° de peticiones con errores de transcripción de pruebas por personal del laboratorio/total peticiones registradas por personal del laboratorio | 1 |
| N.° de peticiones con errores de transcripción de pruebas por personal externo/total peticiones registradas por personal externo | 1 | |
| Tipo de muestra incorrecto | N.° muestras con incorrecto o inapropiado tipo de matriz/n.° total de muestras | 1 |
| N.° muestras con contenedor incorrecto/n.° total de muestras | 1 | |
| Volumen de llenado incorrecto | N.° muestras con volumen insuficiente/n.° total de muestras | 1 |
| N.° muestras con relación muestra-anticoagulante incorrecto/n.° total de muestras con anticoagulante | 1 | |
| Errores de muestras por transporte y conservación | N.° muestras no recibidas/n.° total de muestras | 1 |
| N.° muestras mal conservadas antes de análisis/n.° total de muestras | 1 | |
| N.° muestras dañadas en transporte/n.° muestras transportadas | 1 | |
| N.° muestras transportadas a temperatura incorrecta/n.° total muestras | 1 | |
| N.° muestras con excesivo tiempo de transporte/ n.° total muestras | 1 | |
| Muestras contaminadas | N.° de muestras microbiológicas contaminadas rechazadas/n.° de muestras microbiológicas totales | 1 |
| N.° de muestras contaminadas rechazadas/n.° de muestras no microbiológicas totales | 1 | |
| Muestras hemolizadas | N.° muestras con Hb>0,5g/l detectadas visualmente/n.° muestras revisadas para hemólisis | 1 |
| N.° muestras con Hb>0,5g/l detectadas mediante índices séricos/n.° muestras revisadas para hemólisis | 1 | |
| N.° muestras rechazadas por hemólisis/n.° muestras revisadas para hemólisis | 1 | |
| Muestras coaguladas | N.° muestras coaguladas/n.° muestras con anticoagulante revisadas | 1 |
| Obtención de muestras en tiempo inadecuado | N.° muestras obtenidas en tiempo inadecuado/n.° muestras que requieren tiempo determinado | 2 |
En las recomendaciones finales de este documento no se incluye una propuesta concreta de especificaciones de la calidad para los indicadores de la fase preanalítica por considerar que en el momento actual no están suficientemente estandarizados los métodos para la obtención de los indicadores, lo que puede llevar a sesgos importantes en el cálculo de los mismos.
Garantía externa de la calidad de la fase preanalíticaObjetivos de la participaciónSe recomienda la participación en programas de garantía externa de la calidad en la fase preanalítica como parte del programa de aseguramiento de la calidad de esta fase. La participación permite la evaluación del desempeño actual mediante la comparación con otros laboratorios (estado del arte) y la evolución del nivel de calidad en el tiempo. La participación en estos programas promueve además el establecimiento de un programa de control de calidad interno basado en el cálculo de indicadores que satisface la recomendación de la norma UNE-EN ISO 15189.
El análisis de los resultados del Programa de Garantía Externa de la Calidad Preanalítica de la SEQCML entre los años 2001 y 2013 muestra una evolución favorable, con una reducción de los errores monitorizados en los laboratorios participantes en un control externo de la calidad preanalítica17.
Tipos de programasActualmente, los programas de garantía externa de la calidad de la fase preanalítica pueden diferenciarse en 3 tipos:
- 1)
Recogida de información acerca de procedimientos preanalíticos existentes en el laboratorio a través del envío de cuestionarios o encuestas, como por ejemplo qué criterios de rechazo se aplican a las diferentes muestras y cómo estos se comunican a los médicos solicitantes (Clinical Chemistry EQA Program, Noruega; Quality Control Center, Suiza; WEKAS, Reino Unido; INSTAD, Alemania). Permiten conocer los procedimientos habituales y establecer recomendaciones de consenso.
- 2)
Envío de material de control simulando errores. Por ejemplo, distribución de muestras con alguna sustancia interferente. Es similar a los programas de garantía externa de la calidad analítica convencionales y son muy útiles para controlar las herramientas de detección de especímenes deteriorados (índices séricos) (WEQAS, Reino Unido; SEQCML, España).
- 3)
Registro de errores/incidencias preanalíticas, pudiendo ser indicadores algunos de ellos. El organizador del programa debe suministrar informes que incluyan la evaluación de los resultados del laboratorio comparándolos con los resultados informados por el resto de participantes (CAP, Estados Unidos; KIMMS QA, Australia y Nueva Zelanda; SEQCML, España, así como el MQI del WG-LEPS de la IFCC).
Lo ideal para el laboratorio es participar en los 3 tipos de programas para poder monitorizar un mayor número de errores36.
En el caso de los programas de tipo 1 y 3, la comparación de los resultados de los indicadores entre diferentes países facilitaría la implementación de guías para la armonización/estandarización de diferentes procesos de la fase preanalítica. Sin embargo, para ello es necesario que los datos utilizados para calcular los diferentes indicadores sean los mismos, y un mayor grado de interrelación entre los organizadores.
Recomendaciones1. Se recomienda la implementación de un plan de aseguramiento de la calidad de la fase preanalítica que cumpla con las siguientes condiciones:
- -
Que esté centrado en el paciente, para lo cual se requiere implantar una adecuada gestión del riesgo.
- -
Que cumpla con los requisitos de la norma ISO 15189, tanto en los aspectos de gestión como técnicos.
- -
Que tenga en cuenta las recomendaciones de organismos nacionales e internacionales respecto a la estandarización de procedimientos, armonización de indicadores y establecimiento de especificaciones de la calidad.
2. El control de calidad interno de la fase preanalítica debe basarse en la identificación de riesgos, detección de errores y establecimiento de indicadores. Junto con el mapa de procesos de la fase preanalítica, cada laboratorio debe establecer su mapa de riesgos, mediante la identificación de puntos críticos y modos de fallo habituales. Se recomienda utilizar herramientas de análisis de riesgos proactivas como el análisis modal de fallos y efectos (AMFE) para definir y priorizar los puntos críticos en los que se deben establecer indicadores de la calidad.
3. Para la detección y registro de errores se recomienda que todas las personas que intervengan en cualquiera de las fases del laboratorio tengan capacidad para detectar y registrar los errores producidos. Siempre que sea posible, las incidencias o errores deben ser registrados en el SIL para que puedan ser visualizados a lo largo de todas las etapas y poder actuar en consecuencia, además de conseguir trazabilidad y permitir la posterior explotación para su evaluación.
4. Se recomienda automatizar los procesos preanalíticos para disminuir el número de errores y para mejorar el registro de incidentes y la generación de indicadores del proceso preanalítico.
5. Se recomienda seleccionar e implantar los indicadores comenzando por los de mayor impacto en la seguridad del paciente, teniendo en cuenta los propuestos por los grupos internacionales y programas de intercomparación, ya que proporcionarán una referencia para el establecimiento de las especificaciones de la calidad. Deben de poder ser calculados con recursos razonables, y a poder ser de forma automatizada, para asegurar su fiabilidad y continuidad. Deben validarse y revisarse de forma periódica.
6. Se recomienda la participación en programas de garantía externa de la calidad de la fase preanalítica.
Este documento tiene la conformidad de las tres Sociedades (AEBM-ML, AEFA y SEQC-ML) como Recomendación profesional en el ámbito del Laboratorio Clínico.
