


Existe un número reducido de empresas que han validado procedimientos de medida basados en la cromatografía líquida de alta resolución (de ahora en adelante procedimientos cromatográficos) para el diagnóstico in vitro y con la marca «CE», lo que obliga a los laboratorios clínicos a desarrollarlos y validarlos por sí mismos. La validación de los procedimientos de medida es un requisito de la norma ISO 15189 de acreditación de sistemas de gestión de la calidad en el laboratorio clínico1.
El documento «Validación analítica de los procedimientos de medida del laboratorio clínico»2 de la Comisión de Metrología y Sistemas Analíticos puede servir como guía para la validación de los procedimientos cromatográficos. Sin embargo, debido a la idiosincrasia de estos procedimientos, algunas de las propiedades metrológicas consideradas en este documento han de estudiarse de una manera más exhaustiva y particular3.
Objeto y campo de aplicaciónEl objeto de este documento es describir las propiedades metrológicas a estudiar en el proceso de validación de los procedimientos cromatográficos y cómo debe realizarse su estudio.
El campo de aplicación abarca los procedimientos cromatográficos que permiten la medición de propiedades biológicas correspondientes a escalas racionales (propiedades «cuantitativas»).
Este documento es aplicable a los nuevos procedimientos cromatográficos incorporados al laboratorio clínico y a las modificaciones de los ya implementados, siempre y cuando se considere que tales modificaciones pueden afectar a sus propiedades metrológicas.
En este documento son aplicables los términos y definiciones recogidos en el documento «Vocabulario de términos de metrología para el laboratorio clínico»4, así como los términos relacionados con la cromatografía recogidos en diferentes guías internacionales de la Federación Internacional de Química Pura y Aplicada (IUPAC)5–8, el Instituto para la Normalización de Laboratorios Clínicos (CLSI)9,10, la Agencia Europea del Medicamento (EMA)11 y la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA)12.
Este documento está basado en las recomendaciones generales de la SEQC para la validación de procedimientos de medida2,3,13–23 y en las guías específicas para los procedimientos cromatográficos publicadas por el CLSI9,10, la EMA11 y la FDA12.
Requisitos y propiedades metrológicas a estudiarLa utilización de un procedimiento cromatográfico determinado precisa de un proceso previo de validación que permita comprobar, mediante la provisión de evidencia objetiva, que se han cumplido los requisitos específicos para su utilización prevista1. Por lo tanto, es imprescindible que los laboratorios clínicos establezcan previamente estos requisitos antes de llevar a cabo el proceso de validación. Estos requisitos pueden estar basados en disposiciones legales, ser obtenidos a partir de recomendaciones publicadas por organizaciones científicas o bien, establecidos por el propio laboratorio.
Las principales propiedades metrológicas que deben estudiarse en la validación son:
Exactitud de medidaEn el documento «Recomendaciones para la estimación de la incertidumbre de medida en el laboratorio clínico»13 se describe cómo puede ser estimada la incertidumbre de medida. Adicionalmente, en el documento «Especificaciones para la exactitud de los procedimientos de medida en el laboratorio clínico»14 se detalla cómo establecer los requisitos de exactitud de medida de los procedimientos de medida en el laboratorio clínico. Ambos documentos son aplicables a los procedimientos cromatográficos.
Precisión de medidaLa estimación de la precisión de medida de un procedimiento cromatográfico puede realizarse en base al documento «Estudio de la precisión de los procedimientos de medida en el laboratorio clínico»15.
Las guías cromatográficas recomiendan realizar el estudio de la imprecisión para 4 valores diferentes de la magnitud en estudio: uno correspondiente al límite de cuantificación y, los otros 3, a valores representativos del intervalo de medida. Estas mismas guías especifican que el requisito para la imprecisión (imprecisión máxima permitida) debe ser del 15%, pudiendo ser del 20% para el límite de cuantificación.
Veracidad de medidaPara el estudio de la veracidad, se recomienda aplicar el procedimiento descrito en el documento «Recomendaciones para el estudio de la veracidad de los procedimientos de medida en el laboratorio clínico mediante la utilización de materiales de referencia»16.
Las guías cromatográficas aconsejan que se estudie el sesgo para un valor de la magnitud en estudio correspondiente al límite de cuantificación y a 3 valores representativos del intervalo de medida. Además, indican que el sesgo relativo máximo permitido (en valor absoluto) debe ser del 15%, y del 20% para el límite de cuantificación.
Si se desea realizar el estudio de la veracidad mediante la comparación de procedimientos de medida, puede emplearse el documento «Recomendaciones para el estudio de la veracidad en el laboratorio clínico mediante la comparación de procedimientos de medida»17.
Capacidad de detecciónEl estudio de la capacidad de detección de un procedimiento cromatográfico puede llevarse a cabo siguiendo el documento «Recomendaciones para el estudio de la capacidad de detección de los procedimientos de medida en el laboratorio clínico»18.
El estudio se debe realizar utilizando dos tipos de muestras: un material de blanco (mezcla de muestras de pacientes —se recomienda utilizar un mínimo de 10 muestras— que no contengan ni el analito en estudio ni el patrón interno), y otra con un valor de la magnitud en estudio correspondiente al límite de cuantificación.
Según las guías cromatográficas, la imprecisión y el sesgo relativo máximos permitidos para el límite de cuantificación deber ser del 20%. Adicionalmente, especifican que la relación señal/ruido ha de ser≥511 o≥109,10,12. Esta relación puede estimarse como el cociente entre las medias de las áreas bajo la curva (o alturas) del pico cromatográfico obtenidas en la muestra con valores cercanos al límite de cuantificación y las medias de las áreas bajo la curva obtenidas en el material de blanco.
Intervalo de medidaEn un procedimiento cromatográfico, el intervalo de medida viene dado por el valor del calibrador que presenta un valor cercano al límite de cuantificación (límite inferior del intervalo de medida) y el valor del material de calibración con una mayor concentración (límite superior del intervalo de medida). Habitualmente, en los procedimientos cromatográficos, existe una relación lineal entre las indicaciones (señales analíticas) y el valor de la magnitud biológica3,9–12.
El documento «Procedimiento recomendado para el estudio de la linealidad de los procedimientos de medida en el laboratorio clínico»19, es aplicable a los procedimientos cromatográficos y describe cómo debe realizarse el estudio de validación de la linealidad.
Para realizar las diluciones necesarias en el estudio de la linealidad, las guías cromatográficas indican que se emplee un material de valor elevado con un valor cercano al límite superior del intervalo de medida (p.ej. el material de calibración con el valor más alto de la magnitud en estudio) y, como diluyente, un material de valor bajo con un valor cercano al límite inferior del intervalo de medida (p.ej. el material de calibración con el valor más bajo de la magnitud correspondiente al límite de cuantificación). Las diferentes diluciones han de procesarse por triplicado y, para cada dilución, se ha de calcular la media, la desviación estándar y el coeficiente de variación de los valores obtenidos. También debe calcularse la diferencia relativa porcentual entre los valores obtenidos y su correspondiente valor teórico dividido por el valor teórico, así como la diferencia relativa porcentual media (media de los valores obtenidos menos el valor teórico, dividido por el valor teórico)3.
Los requisitos para la validación de la linealidad son9–12:
- -
El coeficiente de variación obtenido para cada una de las diluciones debe ser ≤ 15%, excepto para la solución a valores cercanos al límite de cuantificación que puede presentar valores ≤ 20%.
- -
Las diferencias relativas porcentuales deben estar comprendidas entre±15%, excepto para la solución a valores cercanos al límite de cuantificación que puede presentar valores comprendidos entre±20%.
En el material suplementario 1 se muestra un ejemplo de estimación del intervalo de medida.
Selectividad y magnitudes influyentesUn procedimiento cromatográfico debe ser capaz de diferenciar el analito en estudio y su patrón interno de otros componentes que puedan encontrarse en la muestra. Para verificar esta propiedad es necesario llevar a cabo un estudio de la selectividad.
Dado que es imposible conocer todos los componentes de una muestra que pueden interferir en un procedimiento cromatográfico, se aconseja estudiar aquellos posibles componentes que tengan una mayor probabilidad de afectar a los resultados. Por ejemplo, diferentes metabolitos del analito y del patrón interno, productos de degradación del analito y del patrón interno que puedan formarse durante la preparación de la muestra y otras sustancias relacionadas con el analito y el patrón interno (sustancias con una estructura química similar o aquellos fármacos más comunes que pueden ser administrados a los pacientes).
Muestras para el estudioLas muestras necesarias para llevar a cabo el estudio de la selectividad son:
- -
Un material de valor bajo con un valor cercano al límite inferior del intervalo de medida (p.ej. el material de calibración con el valor más bajo de la magnitud correspondiente al límite de cuantificación) (LQ).
- -
Muestras de pacientes (un mínimo de 6). Las muestras de pacientes no deben contener el analito en estudio ni su patrón interno, pero sí otros componentes que se sospeche puedan interferir en la medición de la magnitud en estudio. Por ejemplo, en el caso del estudio de magnitudes farmacológicas, las muestras seleccionadas deben contener fármacos diferentes a los que se quiere estudiar o alguno o varios metabolitos del fármaco en estudio.
El material de valor bajo y todas las muestras deben ser distribuidas en alícuotas y almacenadas a -80°C hasta su procesamiento.
Procedimiento y análisis de los resultados- 1)
Procesar el material de valor bajo y las muestras de los pacientes.
- 2)
Obtener los diferentes valores de las señales analíticas para todas las muestras procesadas.
Los requisitos para el estudio de la selectividad de medida son9–12:
- 1)
En el tiempo de retención del analito, los valores de las áreas bajo la curva (o alturas) del pico cromatográfico obtenidos para cada una de las muestras de pacientes han de ser un 20% inferiores al valor del área bajo la curva (o altura) del pico cromatográfico obtenido para la muestra de valor bajo:
- 2)
En el tiempo de retención del patrón interno, los valores de las áreas bajo la curva (o alturas) del pico cromatográfico obtenidos para cada una de las muestras de pacientes han de ser un 5% inferiores al valor del área bajo la curva (o altura) del pico cromatográfico obtenido para la muestra de valor bajo.


En el material suplementario 2 se muestra un ejemplo de cómo llevar a cabo el estudio de la selectividad.
Otras posibles fuentes de interferencia están relacionadas con la hemólisis (hemoglobina), la ictericia (bilirrubina) y la turbidez (triglicéridos), que pueden interferir en los valores obtenidos mediante la HPLC acoplada a la espectrometría de absorción molecular. Para estudiar este tipo de interferencias, se recomienda utilizar el documento «Procedimiento para el estudio de la interferencia por hemólisis, bilirrubina y turbidez y para la verificación de los índices de hemólisis, ictericia y lipidemia»20.
Contaminación por arrastreUno de los estudios que debe realizarse cuando se valida un procedimiento cromatográfico es la contaminación por arrastre o residualidad, dado que la existencia de la misma puede dar lugar a valores falsamente aumentados de la magnitud en estudio.
Muestras para el estudioLas muestras necesarias para realizar el estudio de la contaminación por arrastre son:
- -
Un material de valor bajo con un valor cercano al límite inferior del intervalo de medida (p.ej. el material de calibración con el valor más bajo de la magnitud correspondiente al límite de cuantificación) (LQ).
- -
Un material de blanco. Debe prepararse a partir de una mezcla de muestras de pacientes (se recomienda utilizar un mínimo de 10 muestras) que no contengan ni el analito en estudio ni el patrón interno.
- -
Un material de valor elevado con un valor cercano al límite superior del intervalo de medida (p.ej. el material de calibración con el valor más alto de la magnitud en estudio).
Todos los materiales deben conservarse en condiciones que aseguren la estabilidad de la magnitud durante el tiempo que dure el estudio, por lo que es conveniente distribuirlas en alícuotas y almacenarlas a -80°C.
Procedimiento y análisis de los resultados- 1)
Procesar las diferentes muestras en el siguiente orden:
Material de valor bajo−Material de valor elevado−Material de blanco−Material de blanco−Material de blanco
- 2)
Obtener los diferentes valores de las señales analíticas de cada uno de los materiales procesados.
Los requisitos para el estudio de la contaminación por arrastre son9–12:
- 1)
En el tiempo de retención del analito, los valores de las áreas bajo la curva del pico cromatográfico obtenidos para cada uno de los materiales de blanco deben ser un 20% inferiores al valor del área bajo la curva del pico cromatográfico obtenido para la muestra de valor bajo:
- 2)
En el tiempo de retención del patrón interno, los valores de las áreas bajo la curva del pico cromatográfico obtenidos para cada uno de los materiales de blanco han de ser un 5% inferiores al valor del área bajo la curva del pico cromatográfico obtenido para la muestra de valor bajo:


Si no se cumplen estos requisitos, es probable que exista una contaminación por arrastre. Para evitar esta contaminación, y cuando se procesen muestras de pacientes, deben intercalarse los materiales de blanco entre las mismas.
En el material suplementario 3 se muestra un ejemplo de estudio de la contaminación por arrastre.
EficienciaLa eficiencia de un proceso cromatográfico depende de la recuperación del analito en estudio durante el proceso de extracción de la muestra, y del efecto combinado que diferentes componentes de la muestra pueden ejercer sobre la señal analítica del analito (efecto matriz). El efecto matriz se acentúa en los procedimientos de medida basados en la HPLC acoplada a la espectrometría de masas (HPLC-MS) o a la espectrometría de masas en tándem (HPLC-MS/MS), debido al efecto que pueden producir en la señal analítica los componentes al coeluir con el analito durante el proceso de ionización de la muestra.
La recuperación se calcula a partir del cociente entre las señales que proporciona un analito que ha sido añadido antes y después de un proceso de preparación o extracción de muestras sobre muestras de pacientes que no contienen el analito (muestras de blanco). En cambio, el efecto matriz se calcula a partir del cociente entre las señales obtenidas del analito cuando este es añadido después del proceso de extracción de muestras de blanco y en un disolvente apropiado (habitualmente la fase móvil).
Muestras para el estudioLas muestras necesarias para llevar a cabo estos estudios son:
- -
Un mínimo de 6 muestras de blanco.
- -
Nota: si el analito es un componente biológico, como muestras de blanco, es recomendable utilizar muestras de pacientes que presenten un valor de la magnitud en estudio lo más bajo posible.
- -
Un extracto de cada una de las muestras de blanco de pacientes, obtenido mediante el proceso de extracción cromatográfico empleado.
- -
Materiales de pureza conocida del analito y del patrón interno.
Soluciones de trabajo del analito preparadas en muestras de blanco (antes del proceso de extracción; preextracción)
- 1)
A partir del material de pureza conocida del analito, preparar una solución primaria del analito en un disolvente apropiado (agua, metanol, acetonitrilo, dimetilsulfóxido, dependiendo de la solubilidad del analito).
- 2)
A partir de la solución primaria, y para cada muestra de blanco, preparar tres soluciones de trabajo con diferentes concentraciones de analito. Dos de ellas con valores patológicos de la magnitud en estudio (una por debajo y otra por encima del intervalo de referencia, intervalo terapéutico o valor de decisión clínica), y la tercera, con valores fisiológicos.
Nota: en algunos casos, los diferentes disolventes orgánicos utilizados en la preparación de la solución primaria pueden provocar la precipitación de las muestras de blanco, por lo que es preferible realizar distintas soluciones secundarias, en agua calidad HPLC o HPLC-MS, y preparar las soluciones de trabajo a partir de estas soluciones secundarias y las muestras de blanco.
Soluciones de trabajo del analito preparadas en el extracto de las muestras de blanco (después del proceso de extracción; postextracción)
Realizar el mismo procedimiento del apartado anterior pero utilizando los extractos de las muestras de blanco en lugar de las muestras. Los valores para estas soluciones deben ser los mismos que los de las soluciones preparadas en el apartado anterior.Soluciones de trabajo del analito en un disolvente apropiado (fase móvil)
Realizar el mismo procedimiento del apartado anterior pero utilizando la fase móvil como disolvente en lugar de las muestras de blanco de pacientes. Los valores para estas soluciones deben ser los mismos que los de las soluciones preparadas en los apartados anteriores.Soluciones de trabajo del patrón interno
Preparar soluciones idénticas a las descritas en los apartados anteriores, pero utilizando un material de elevada pureza del patrón interno en lugar del analito.
Procedimiento y análisis de los resultados- 1)
Procesar, aleatoriamente, por triplicado y en una misma serie, todas las soluciones de trabajo.
Nota: no se debe realizar el proceso de extracción para las soluciones de trabajo preparadas en el extracto de las muestras de blanco y han de procesarse directamente en el sistema cromatográfico.
- 2)
Obtener los diferentes valores de las señales analíticas (áreas bajo la curva o alturas del pico cromatográfico) para todas las soluciones procesadas.
- 3)
Para cada triplicado de cada solución de trabajo, calcular el porcentaje de recuperación (RE) y el porcentaje del factor de matriz (MF), empleando las siguientes ecuaciones:
- 4)
Para conocer si el patrón interno es capaz de compensar la pérdida de analito durante el proceso de extracción de la muestra, así como el posible efecto matriz, deben calcularse el porcentaje de recuperación normalizado (n-RE) y el porcentaje de factor matriz normalizado (n-ME), empleando las siguientes fórmulas:
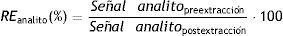





Los requisitos para el estudio de la recuperación y el efecto matriz son9–12:
- 1)
Para cada solución de trabajo, el coeficiente de variación entre replicados debe ser ≤ 15%.
- 2)
Los diferentes valores de recuperación y del factor matriz normalizados deben ser reproducibles e independientes del valor de la solución de trabajo. Es decir, que los valores del porcentaje de recuperación y del factor matriz conjuntos o globales normalizados (media de todos los valores de recuperación o del factor matriz del analito normalizados estudiados) han de presentar un coeficiente de variación ≤ 15% y estar comprendidos dentro del intervalo 100%±15% ([85-115] %).
Si no se cumplen estos requisitos es necesario seleccionar otro patrón interno y realizar nuevamente el estudio, dado que el patrón interno escogido no es capaz de compensar ni la pérdida del analito durante el proceso de extracción de la muestra ni el efecto matriz existente.
Adicionalmente, aunque no es obligatorio su cumplimiento y para cada una de las soluciones de trabajo, los valores de recuperación y de factor de matriz para el analito y el patrón interno deben ser similares entre sí y estar comprendidos entre el 85 y el 115%.
En los materiales suplementarios 4 y 5 se muestran ejemplos de cómo estudiar la recuperación y el efecto matriz, respectivamente.
Integridad de las dilucionesLas guías cromatográficas recomiendan que se lleve a cabo un estudio de la integridad de las diluciones que se realicen cuando el valor de la magnitud sea superior al límite superior del intervalo de medida.
Muestras para el estudioLas muestras necesarias para llevar a cabo el estudio de la integridad de las diluciones son:
- –
Un material de blanco. Se prepara a partir de una mezcla de muestras de pacientes (se recomienda utilizar un mínimo de 10 muestras) que no contengan ni el analito en estudio ni el patrón interno.
- –
Un material de pureza conocida del analito.
- –
Un material de valor elevado. Se trata de un material que contiene el analito en estudio con un valor de la magnitud biológica por encima del límite superior del intervalo de medida previamente establecido. Este material se prepara como sigue:
- 1)
A partir del material de pureza conocida del analito, preparar una solución primaria del analito en un disolvente apropiado (agua, metanol, acetonitrilo, dimetilsulfóxido, dependiendo de la solubilidad del analito).
- 2)
A partir de la solución primaria y el material de blanco, preparar una solución de trabajo con un valor de la magnitud en estudio 2 veces el valor del límite superior del intervalo de medida.
Nota: en algunos casos, los diferentes disolventes orgánicos utilizados en la preparación de la solución primaria pueden provocar la precipitación del material de blanco empleado, por lo que es preferible realizar una solución secundaria, en agua calidad HPLC o HPLC-MS, y preparar la solución de trabajo a partir de esta solución secundaria y el material de blanco.
- -
Realizar una dilución 1/3, 1/5 y 1/10 de la solución de trabajo empleando como disolvente el material de blanco.
- -
Procesar, aleatoriamente y en una misma serie, cada una de las diluciones un mínimo de seis veces.
Los requisitos para el estudio de la integridad de las diluciones son:
- -
Para cada dilución, el coeficiente de variación obtenido entre replicados debe ser ≤ 15%.
- -
Para cada dilución, el porcentaje de desviación (diferencia porcentual entre la media de los valores obtenidos y su correspondiente valor teórico dividido por el valor teórico) ha de estar comprendido entre±15%.
En el material suplementario 6 se muestra un ejemplo de cómo realizar el estudio de la integridad de las diluciones.
EstabilidadLas guías cromatográficas especifican que cuando se valida un procedimiento cromatográfico que ha elaborado el propio laboratorio deben llevarse a cabo distintos estudios de estabilidad para asegurar que, durante la preparación de las muestras y en el procesamiento de las mismas así como en las condiciones de almacenamiento usadas, no se ven afectados los valores de las magnitudes. En general, los estudios de la estabilidad se realizan comparando los valores obtenidos en diferentes tipos de muestras que contienen el analito almacenadas durante distintos períodos de tiempo y en diferentes condiciones. Por otro lado, las condiciones en que se realizarán los estudios de estabilidad —las matrices utilizadas, los recipientes de muestra que se emplearán, las condiciones y el tiempo de almacenamiento, entre otros— deben ser similares a las que se utilizarán cuando se trabaje con muestras de pacientes.
Los diferentes estudios de estabilidad que se recomiendan realizar son:
- -
La estabilidad en las diferentes soluciones primarias, secundarias y de trabajo preparadas para el analito y para el patrón interno, bajo las condiciones en que se almacenarán las mismas.
- -
La estabilidad a corto plazo en la matriz en estudio a temperatura ambiente o a la temperatura de procesamiento de las muestras.
- -
La estabilidad a largo plazo en la matriz en estudio a una temperatura de -20°C o -80°C.
- -
El número de ciclos de congelación-descongelación en la matriz en estudio.
- -
La estabilidad en muestras de pacientes a temperatura ambiente o bajo las condiciones de almacenamiento de las mismas.
- -
La estabilidad en el muestreador del procedimiento cromatográfico para el extracto de la matriz en estudio, a temperatura ambiente o bajo las condiciones de almacenamiento de las mismas.
Las muestras necesarias para llevar a cabo los estudios de estabilidad son:
- -
Las soluciones primarias y secundarias del analito y del patrón interno.
- -
Una mezcla de muestras de pacientes que no contenga el analito (material de blanco) a la que se le ha añadido diferentes cantidades del analito a partir de un material de pureza conocida (por ejemplo, pueden emplearse los materiales de control que se utilizan para comprobar el correcto funcionamiento del sistema cromatográfico).
- -
Extractos de la mezcla de muestras de pacientes que no contengan el analito (material de blanco) a los que se les ha añadido una cantidad determinada del analito a partir de un material de pureza conocida. Estos extractos se obtienen después de realizar el proceso de extracción cromatográfico (por ejemplo, pueden emplearse los extractos de materiales de control).
- -
Muestras de pacientes (un mínimo de 20) que contengan el analito.
Estudio de la estabilidad de las soluciones primarias y secundarias
Para llevar a cabo este estudio:
- 1)
Decidir el periodo de tiempo y las condiciones de almacenamiento de las soluciones.
- 2)
Almacenar las diferentes soluciones durante el periodo de tiempo y bajo las condiciones previamente escogidas.
- 3)
Una vez transcurrido el periodo de tiempo de almacenamiento de las soluciones, después de atemperarlas y agitarlas, procesarlas de manera aleatoria y en una misma serie un mínimo de 6 veces.
- 4)
Para cada solución, calcular la diferencia relativa porcentual (PD) utilizando la siguiente fórmula:
donde Xi es cada uno de los valores obtenidos en la solución, Y es el valor teórico o nominal de la solución y n es el número de veces que se ha procesado la solución (en este caso, n=6).

Se considera que la magnitud es estable si la diferencia relativa porcentual está comprendida entre±15%.
Estudio de la estabilidad a corto y largo plazo
Para realizar estos estudios:
- 1)
Decidir el período de tiempo y las condiciones de almacenamiento para los materiales de control.
- 2)
Almacenar los materiales de control, a temperatura ambiente (para el estudio a corto plazo) y a -20°C o -80°C (para el estudio a largo plazo) durante el periodo de tiempo escogido.
- 3)
Una vez transcurrido el periodo de tiempo de almacenamiento de los materiales de control, después de atemperarlos y agitarlos, procesarlos aleatoriamente y en una misma serie un mínimo de 6 veces.
- 4)
Para cada material de control, calcular la diferencia relativa porcentual (PD) utilizando la siguiente fórmula:
donde Xi es cada uno de los valores obtenidos en el material de control, Y es el valor teórico o nominal del material de control y n es el número de veces que se han procesado (en este caso, n=6).
Se considera que la magnitud es estable si la diferencia relativa porcentual está comprendida entre±15%.
Estudio del número de ciclos de congelación-descongelación
Para llevar a cabo este estudio:
- 1)
Preparar los materiales de control y almacenarlos a -20°C o -80°C durante un periodo de tiempo mínimo de 12 h.
- 2)
Una vez transcurrido el tiempo, después de atemperarlos y agitarlos, procesarlos aleatoriamente y en una misma serie un mínimo de 6 veces.
- 3)
Para cada material de control, calcular la diferencia relativa porcentual (PD) utilizando la siguiente fórmula:
donde Xi es cada uno de los valores obtenidos en el material de control, Y es el valor teórico o nominal del material de control y n es el número de veces que se han procesado (en este caso, n=6). Se considera que la magnitud es estable si la PD está comprendida entre±15%.
- 4)
Volver a almacenar los materiales de control a la temperatura de congelación durante otro periodo de tiempo mínimo de 12 h.
- 5)
Repetir los puntos 2), 3) y 4) hasta que el valor de la PD sobrepase el intervalo±15%. El número de ciclos de congelación-descongelación corresponderá al número de veces que se ha realizado este estudio de estabilidad y la PD ha estado comprendida entre±15%.
Estudio de la estabilidad en muestras de pacientes
La SEQC ha publicado diversos documentos21,22 que especifican cómo ha de realizarse el estudio de la estabilidad de las magnitudes biológicas, así como los criterios para establecer el límite de estabilidad23. Este documento es transferible a las magnitudes biológicas que se miden mediante procedimientos cromatográficos.
Estudio de la estabilidad en el muestreador del sistema cromatográfico
Para realizar este estudio:
- 1)
Decidir el período de tiempo y las condiciones de almacenamiento que se pretenden estudiar.
- 2)
Realizar el procedimiento de extracción para los materiales de control y almacenar los extractos, dentro del muestreador del sistema cromatográfico, durante el periodo de tiempo y bajo las condiciones de temperatura previamente escogidas.
- 3)
Una vez transcurrido el periodo de tiempo de almacenamiento de los extractos de los materiales de control, después de atemperarlos y agitarlos, se procesan aleatoriamente y en una misma serie un mínimo de 6 veces.
- 4)
Para cada extracto de un material de control determinado, calcular la PD utilizando la siguiente fórmula:
donde Xi es cada uno de los valores obtenidos en el extracto del material de control, Y es el valor teórico o nominal del extracto del material de control y n es el número de veces que se han procesado (en este caso, n=6).
Se considera que la magnitud es estable si la diferencia relativa porcentual está comprendida entre±15%.
En el material suplementario 7 se muestra un ejemplo de los distintos estudios de estabilidad.
