



El carcinoma mioepitelial es de aparición rara en la glándula submaxilar. Presentamos un caso clínico y una revisión de la literatura que nos permiten establecer un diagnóstico diferencial y un tratamiento óptimo de este tumor. La histología y la inmunohistoquímica desempeñan un papel importante en el diagnóstico definitivo.
Myoepithelial carcinomas or malignant myoepitheliomas are considered an uncommon salivary gland tumour with a predilection for the parotid gland. A case report is presented, along with a literature review in order to investigate the biological behaviour and proper management of myoepithelial carcinomas. Clinicopathological and immunohistochemical features are shown to achieve a better understanding this entity and to prevent their easy confusion with many other tumours.
El carcinoma mioepitelial o mioepitelioma maligno es una neoplasia de las glándulas salivales poco frecuente, compuesta por células tumorales con diferenciación mioepitelial. El término mioepitelioma se refiere a cualquier neoplasia compuesta predominantemente por células mioepiteliales, distinguiéndose mioepiteliomas benignos y malignos. La mayoría de los mioepiteliomas descritos en la literatura son benignos. En 1943, Sheldon fue el primero en descubrir estos tumores mioepiteliales cuando identificó 3 de ellos en una revisión de 57 tumores mixtos de las glándulas salivales1. El componente maligno (mioepitelioma maligno) fue descrito mucho más tarde por Stromayer et al. en 19752.
Esta neoplasia maligna representa el 0,4-0,6% de todas las de glándulas salivales3 y el 1,2-1,5% de todos los carcinomas2. Afecta habitualmente a pacientes de mediana y avanzada edad (media de edad 55 años, rango 14-86 años)2,4 y no muestra predominio por ningún sexo. Surge predominantemente de las glándulas salivales mayores, habitualmente en la glándula parótida, y muestra un patrón de crecimiento infiltrativo hacia los tejidos colindantes. La afectación de las glándulas salivales menores es rara, habiéndose descrito 69 casos en la literatura inglesa. Los casos de afectación de estructuras intraorales (paladar, lengua) son aún más raros.
Los criterios diagnósticos aceptados actualmente para el carcinoma mioepitelial son la diferenciación exclusiva mioepitelial (morfológica e inmunohistoquímica) y el carácter maligno con infiltración de la glándula salival y los tejidos adyacentes5,6.
Presentamos un caso de carcinoma mioepitelial que afecta a la glándula submaxilar y el tratamiento realizado. La rareza del tumor, junto a su gran capacidad de recurrencia y metástasis, sugiere un análisis exhaustivo del caso clínico y de la literatura, para enfatizar en los aspectos clínicos, anatomopatológicos, inmunohistoquímicos y tratamientos óptimos, en relación con otros tumores de localización similar.
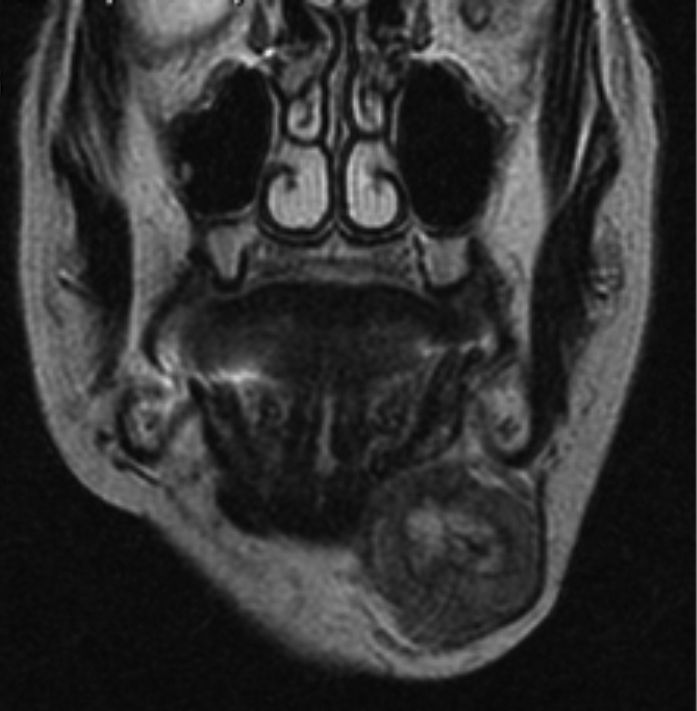
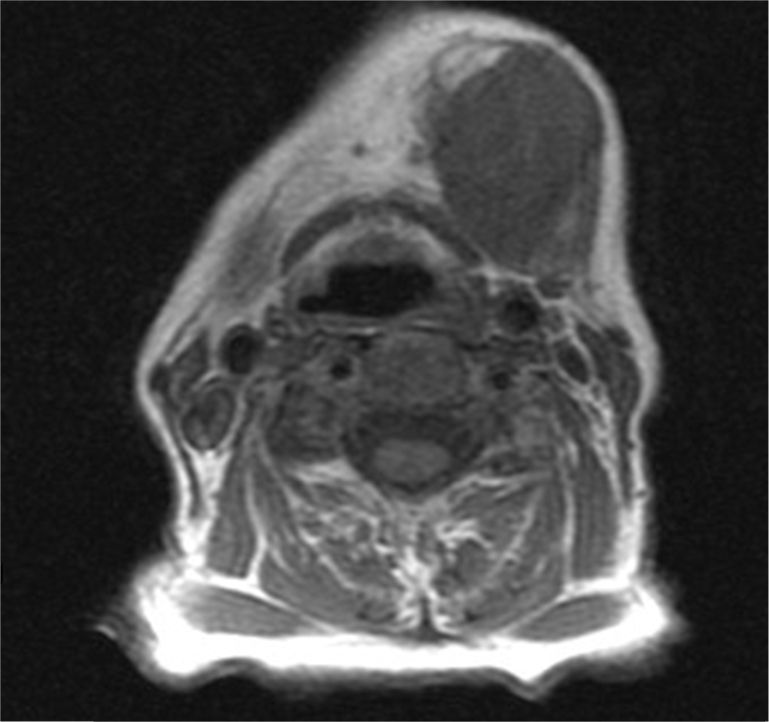
Caso clínicoMujer de 80 años de edad, que acude a las consultas por presentar una tumoración indolora submandibular izquierda de 2 meses de evolución y que ha experimentado un crecimiento progresivo y un aumento de la consistencia. No se acompaña de síndrome constitucional. La paciente presenta litiasis renal e hipertensión arterial como únicos antecedentes. En la exploración física destaca una masa submandibular izquierda de aproximadamente 4cm de diámetro, de consistencia dura, adherida a planos profundos, que no provoca dolor a la palpación. No se observan alteraciones en la movilidad cervical ni en la expresión de la mímica facial. En la exploración intraoral no hay inflamación del suelo de la boca ni del conducto de Wharton. Se procede a la realización de una punción-aspiración con aguja fina (PAAF) guiada por ecografía que se informa como carcinoma de naturaleza indeterminada. Se solicita una RM cuyo informe es compatible con una masa submaxilar izquierda heterogénea, con necrosis interna, de 3×4×3cm, que impronta en el suelo de la boca, sin signos concluyentes de infiltración de la musculatura suprahioidea y que respeta la cortical ósea mandibular (figs. 1 y 2).


Con el diagnóstico de tumoración maligna de la glándula submaxilar, se realiza una disección cervical funcional izquierda tipo iii, incluyendo el nervio hipogloso, el músculo digástrico y la rama marginal del nervio facial izquierdo, con un fresado de la cara medial mandibular izquierda.
El análisis histológico de la pieza muestra una proliferación neoplásica de límites infiltrantes, que crece formando nidos y cordones, constituida por células de citoplasmas amplios y morfología plasmocitoide y epitelioide. Los núcleos son atípicos con moderado pleomorfismo, nucléolo prominente y 11 mitosis/10 campos de gran aumento. Las células se disponen en el seno de un estroma hialinizado. Se aprecian extensas zonas de necrosis y focos de diferenciación escamosa. No se observan estructuras ductales. El estudio inmunohistoquímico realizado demuestra positividad de las células neoplásicas para CKAE1/AE3, actina, calponina, p63, proteína S-100 y GFAP, siendo negativas para CEA. El índice proliferativo (Ki67) es del 25%. El tumor está totalmente resecado y la distancia al margen de resección más próximo es de menos de 1mm. Los ganglios linfáticos son negativos para malignidad. Este informe es compatible con el diagnóstico de carcinoma mioepitelial de glándula submaxilar pT3-N0, MX.
Se decide, tras un consenso multidisciplinar, la aplicación de radioterapia postoperatoria. La dosis total de radioterapia recibida es de 60Gy, fraccionado en 30 sesiones.
La paciente está en seguimiento en consultas externas después de 3 años de la intervención quirúrgica y, tras la realización de RM y tomografía computarizada seriadas de control, no se visualizan signos de recidiva tumoral ni metástasis.
DiscusiónLas células mioepiteliales se describen por primera vez en el año 1898 por Zimmerman7. Se definen como unas células planas y contráctiles que rodean las unidades glandulares, acinares y ductales de varios órganos, como las glándulas salivales y la mama. A diferencia de otras células contráctiles, se dice que se originan del ectodermo, como el sistema glandular. La contracción de estas células ayudaría a impulsar los productos de secreción hacia los conductos excretores mayores7.
Los carcinomas de glándulas salivales con diferenciación mioepitelial (carcinoma mioepitelial o mioepitelioma maligno) son de aparición muy rara y fueron descritos más tardíamente que la variante benigna. Sometidas a múltiples controversias, las células mioepiteliales continúan siendo, actualmente, objeto de estudio por sus funciones poco definidas y estructuras complejas. En 1985, Barnes et al. revisan la literatura de todos los mioepiteliomas de cabeza y cuello, describiendo solo 3 casos de carcinoma mioepitelial. Con 2 publicaciones, en 1989 y 1995, Dardick et al. presentan las diferentes variantes de mioepiteliomas con las respectivas características y criterios diagnósticos1,7. Como consecuencia de la publicación de más casos, los mioepiteliomas malignos fueron incluidos dentro de la segunda edición de la clasificación histológica de tumores de glándulas salivales de la Organización Mundial de la Salud, en 19911,3,7,8.
Estos tumores representan menos del 1% del total y solamente 9 series de casos evaluando las características clinicopatológicas han sido publicadas hasta la fecha. En el año 2000, se realiza una revisión de la literatura mundial y se observan aproximadamente 75 casos de carcinoma mioepitelial7. Se distinguen de los benignos por su crecimiento infiltrativo y destructivo, pleomorfismo celular y actividad mitótica1. En el caso presentado, la diferenciación mioepitelial se establece por unos criterios histológicos e inmunohistoquímicos.
Los mioepiteliomas malignos surgen habitualmente de novo o como un carcinoma secundario a un adenoma pleomorfo o un mioepitelioma benigno preexistente2,4,6,9. El caso presentado evoca un mioepitelioma maligno surgido de novo en una glándula salival normal. Los tumores de novo tienen tendencia a la agresividad clínica y radiológica, con un tiempo de evolución corto. Los tumores surgidos de adenomas pleomorfos o de tumores benignos se sospechan cuando existe una historia personal larga de un tumor benigno salival con un crecimiento repentino rápido o cuando existen múltiples recurrencias en un adenoma pleomorfo preexistente con o sin metástasis ganglionar6,7. Se sabe que los tumores malignos que surgen de un adenoma pleomorfo son muy agresivos, pero esta característica no ha sido confirmada para el mioepitelioma maligno10.
Clínicamente, se comporta como una masa con tendencia a invadir y destruir los tejidos adyacentes. La duración de los síntomas antes del diagnóstico varía desde un mes hasta 3 años. Algunos autores aseguran que el tumor mantiene un tamaño pequeño durante un cierto periodo y experimentan posteriormente un crecimiento muy rápido1. Existe evidencia de malignidad cuando se objetiva una larga evolución, un crecimiento rápido, presencia de dolor, signos de parálisis facial, una consistencia pétrea, una adhesión a planos profundos y presencia de adenopatías patológicas. En nuestro caso, ningún síntoma nos orienta hacia un carácter maligno de la tumoración, siendo imperativa, por lo tanto, la ayuda de las pruebas radiológicas y citológicas.
La PAAF es habitualmente una herramienta sensible y específica para el diagnóstico de las tumoraciones11. En un estudio Behzatoglu et al. concluyen que la sensibilidad y la especificidad de la PAAF en el diagnóstico de masas parotídeas es del 91 y el 98%, respectivamente12. Igualmente aseguran que ciertas entidades, como el mioepitelioma maligno, son de difícil identificación con esta técnica6. Existen pocos estudios en la literatura que avalen su fiabilidad en esta patología, siendo la mayoría de ellos casos clínicos que presentan resultados contradictorios. En 31 casos publicados por Kumar et al. (7 casos)13, Chhieng et al. (4 casos)8, Miliauskas y Orell (5 casos)14 y Khademi et al. (15 casos)15, ninguno es diagnosticado en el preoperatorio de neoplasia mioepitelial con PAAF8.
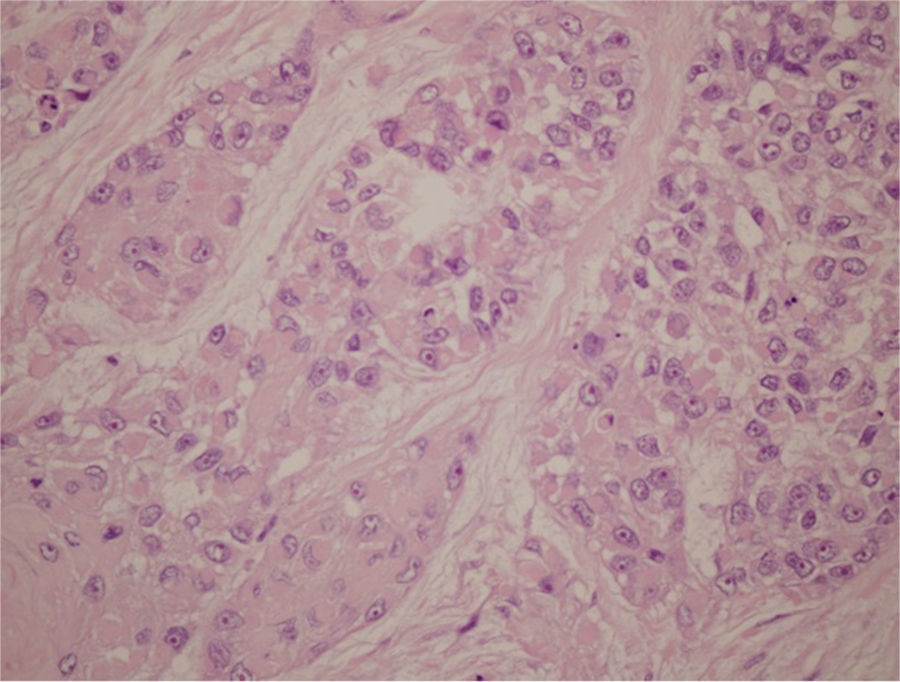
Al igual que el mioepitelioma benigno, la contrapartida maligna se caracteriza por una gran heterogeneidad morfológica, coexistiendo frecuentemente varios tipos celulares (células fusiformes, epitelioides, plasmacitoides y claras) y distintos patrones arquitecturales (en sábana, multinodular, nidos y cordones) (fig. 3)8. Pueden observarse una abundante matriz hialina y/o mixoide y unos cambios metaplásicos (metaplasia escamosa, sebácea y condroide) focales. El componente hialino representa elementos relacionados con la membrana basal debido a la capacidad de las células mioepiteliales de producir matriz. En la serie de 51 casos de Kane y Bagwan, se observa una combinación de 2 o más tipos celulares en 12 casos (6 de ellos con células epitelioides y plasmocitoides) y matriz hialina en 26 casos, rasgos que también se evidencian en la presente lesión7. Uno de los temas de controversia en estos tumores es la presencia de estructuras ductales. Aunque por definición estos tumores solo tienen diferenciación mioepitelial, pero no ductal ni acinar, en algunos casos pueden observarse escasas estructuras ductales. Sin embargo, algunos autores no admiten la formación de ductos y reservan el término de carcinoma mioepitelial para aquellos tumores con exclusiva diferenciación mioepitelial. Según estos autores, un carcinoma compuesto predominantemente por células claras mioepiteliales con escasos túbulos epiteliales se denominaría carcinoma epi-mioepitelial16.
: nidos de células plasmocitoides y epitelioides con núcleos atípicos.")
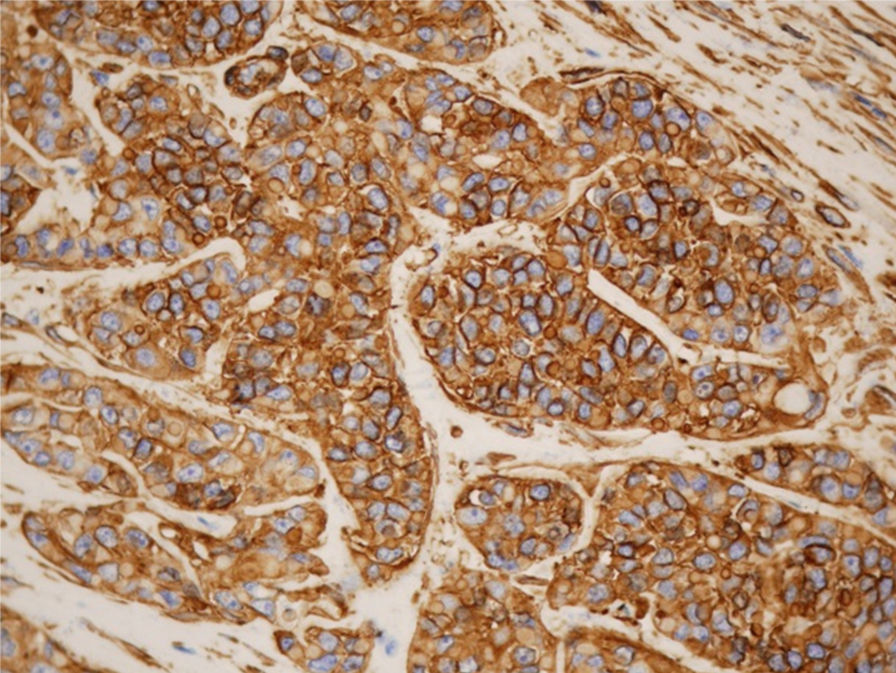
Desde el punto de vista inmunohistoquímico, la expresión en las células tumorales de citoqueratinas (pancitoqueratinas, CAM 5.5 y 34BE12), actina muscular específica, proteína S100, calponina, p63, GFAP y vicentina confirma la diferenciación mioepitelial2,3,7,9,10. La calponina es un anticuerpo de músculo liso que es claramente sensible y específico de diferenciación mioepitelial en tumores de glándula salival (fig. 4). Los marcadores de diferenciación luminal (tubular), especialmente el CEA, son negativos en el carcinoma mioepitelial1,8.
: células neoplásicas expresan calponina en sus citoplasmas.")
Los criterios de malignidad han sido siempre un motivo de discusión. Tradicionalmente, los rasgos histológicos que ayudan a distinguir entre un mioepitelioma maligno y benigno incluyen la atipia citológica, la infiltración y el índice mitótico. Según el artículo de Savera et al., la infiltración tumoral en partes blandas es el rasgo más importante y debería ser un requisito mínimo para el carcinoma mioepitelial1,8. Incluye también otras características, como las mitosis frecuentes y la necrosis, pero da menor importancia a la atipia citológica. Recientemente, se ha indicado que más de 7 mitosis por 10 campos de gran aumento o un índice proliferativo mediante inmunotinción con Ki67 superior al 10% es diagnóstico de malignidad1,8. En esta serie extensa de Savera et al., el 40% de los tumores son clasificados como de alto grado y el 60% de bajo grado1. Los carcinomas mioepiteliales de alto grado muestran marcado pleomorfismo nuclear, extensas zonas de necrosis y más de 4 mitosis por campo. Los de bajo grado presentan ligera variación del tamaño nuclear y cromatina homogéneamente distribuida.
En el diagnóstico diferencial (tabla 1) del carcinoma mioepitelial se incluyen numerosas entidades, dependiendo del tipo celular predominante. En los que predominan las células epitelioides pueden plantearse problemas de diagnóstico diferencial con otras neoplasias de glándula salival, especialmente con el carcinoma adenoide quístico, el adenocarcinoma polimorfo de bajo grado, el carcinoma epi-mioepitelial o el adenocarcinoma NOS. La presencia de estructuras tubulares y/o de células luminales, junto con la demostración de diferenciación luminal inmunohistoquímicamente, con CEA y EMA, favorecen el diagnóstico de carcinoma adenoide quístico, carcinoma epi-mioepitelial o adenocarcinoma. Cuando predominan las células plasmocitoides, pueden confundirse con plasmocitomas o melanomas. Los de células fusiformes pueden confundirse con tumores de vainas nerviosas, leiomiosarcomas, sarcomas sinoviales, fibrohistiocitomas, fibrosarcomas y carcinomas escamosos sarcomatoides. En casos de tumores con morfología de células claras17, pueden plantearse problemas de diagnóstico diferencial con carcinoma de células claras NOS, carcinoma epi-mioepitelial, carcinoma mucoepidermoide y metástasis de un carcinoma renal. Para el patólogo, la combinación de los hallazgos morfológicos, como el patrón bifásico con diferenciación luminal en el carcinoma epi-mioepitelial, la presencia de pigmento melánico en el melanoma, las áreas A y B de Antoni en el schwannoma y el componente escamoso in situ en el carcinoma sarcomatoide, junto con el estudio inmunohistoquímico resultan definitivos para la correcta filiación1,16.
Diagnóstico diferencial histológicoa
| Células epitelioides | Carcinoma adenoide quísticoAdenocarcinoma polimorfo de bajo gradoCarcinoma epi-mioepitelialAdenocarcinoma NOS |
| Células luminales y estructuras tubulares | Carcinoma adenoide quísticoCarcinoma epi-mioepitelialAdenocarcinoma |
| Células plasmocitoides | PlasmocitomasMelanomas |
| Células fusiformes | Tumores de vainas nerviosas3LeiomiosarcomasSarcomas sinovialesFibrohistiocitomasFibrosarcomasCarcinomas escamosos sarcomatoides |
| Células claras17 | Carcinoma de células claras NOS3Carcinoma epi-mioepitelialCarcinoma mucoepidermoideMetástasis de un carcinoma renal |
En cuanto al tratamiento, la cirugía con amplios márgenes de seguridad ha sido propuesta como el más efectivo en el manejo de los carcinomas mioepiteliales2,18. La disección cervical está indicada en los casos de invasión linfática cervical objetivada por la exploración física y las pruebas radiológicas. Las utilidades y las indicaciones de la radioterapia y la quimioterapia no están definidas todavía y son controvertidas1,3,6,16. En nuestro caso, al haber obtenido un resultado positivo de una tumoración maligna indeterminada, se propuso una disección ganglionar asociada a la exéresis del tumor.
El tumor presenta un índice alto de metástasis a distancia pero bajo de invasión metastásica ganglionar cervical. Existe una serie de casos documentados con metástasis en el pulmón y la pleura, ganglios linfáticos inguinales, hígado, médula espinal lumbar, peritoneo, cerebro y costillas4,16. Las metástasis cutáneas son extremadamente raras, habiéndose descrito solo un caso en la literatura, en forma de nódulos cutáneos indurados en manos, tronco y piernas19. En la revisión reciente de 51 casos de Kane y Bagwan, relacionan los siguientes factores con una probabilidad alta de metástasis: tumores con más del 50% de necrosis, márgenes positivos, índice mitótico alto (más de 4 por 10 HPF), un índice Ki-67 entre 4-10%, atipias celulares y patrón celular fusiforme7. Jiang et al. concluyen en su último estudio que la recidiva y las metástasis ocurren más frecuentemente en pacientes Ki-67 y p-63 positivos, y que el pronóstico más ínfimo se relaciona con la sobreexpresión de p6320.
Por otro lado, Savera et al., aseguran en su artículo que no existe ningún criterio histológico que se relacione con el comportamiento clínico del tumor y, por lo tanto, con el pronóstico del mismo. De los 25 pacientes revisados, 10 presentan recidivas. De los pacientes seguidos (solo 17), 8 presentaron metástasis, 6 con un tumor de alto grado y 2 de bajo grado1. Se sabe que un tumor con marcadores histológicos de malignidad (atipia celular, mitosis y necrosis) actúa de manera agresiva, pero se conocen igualmente casos con comportamiento inherente pese a la histología adversa1. Ocasionalmente, se han descrito tumores de bajo grado de malignidad y una actividad mitótica mínima que han metastatizado. Nagao et al. presentan, en 1998, una revisión de 10 casos de carcinoma mioepitelial y 3 de ellos con un perfil agresivo (índice mitótico alto y necrosis) tienen registrada una larga supervivencia: 2 se mantienen libres de enfermedad pasados 8 y 11 de años de seguimiento, mientras que el tercero fallece a los 15 años de la enfermedad21. Di Palma y Guzzo, en 1993, publican sus resultados sobre 10 pacientes: 2 de los pacientes seguían vivos a los 4 años, uno murió de la enfermedad a los 35 años y el cuarto paciente no tiene evidencia de recidiva a los 37 años10. Sin embargo, un caso de mioepitelioma maligno de Ibrahim et al. de 1991 muere a los 3 meses de la cirugía por metástasis hepáticas; no había signos de agresividad histológica. Un caso similar publicado por Hsiao et al. de un carcinoma de bajo grado de malignidad desarrolla metástasis cerebrales y muere a los 5 meses de la cirugía22.
Kane y Bagwan muestran recidiva tumoral en 18 de 44 casos, 5 de ellos con recurrencia múltiple7. Así mismo, relacionan la recidiva con el tipo de células tumorales (morfología estrellada), la existencia de invasión ósea o perineural y el tamaño tumoral (mayor de 6cm)7. Un correcto seguimiento de los pacientes engloba un periodo mínimo de 24 meses. En nuestra paciente, tras un seguimiento de 72 meses, no se observan signos de recurrencia de la enfermedad.
ConclusionesLas características para recordar de este tumor maligno son: predilección parotídea, masa en general de crecimiento rápido con extensión a los tejidos adyacentes, índice alto de metástasis a distancia, recurrencia frecuente después de la exéresis quirúrgica y pronóstico impredecible.
El conocimiento de la histología y la inmunohistoquímica es importante para distinguir esta neoplasia de otras similares y afinar el tratamiento óptimo. La cirugía ha demostrado ser el tratamiento de elección, siendo todavía controvertidas la radioterapia y la quimioterapia. El pronóstico de la enfermedad es incierto y no parecen dilucidarse criterios comunes de mal pronóstico.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesNo existen.
