


El término displasia fibrosa hace referencia a un conjunto de lesiones óseas benignas que se caracterizan por la sustitución del tejido óseo normal por tejido conectivo. Se presenta el caso de una paciente afectada de displasia fibrosa poliostótica de predominio maxilar tratada de forma conservadora con bisfosfonatos
The term fibrous dysplasia refers to a variety of bony diseases characterized by the substituion of the bone by abnormal connective tissue. A case report of patient affected by a polyostotic form of fibrous dysplasia with an uneven evolution of its disease after being treated with pamidronate is presented.
El término displasia fibrosa hace referencia a un conjunto de lesiones óseas benignas que se caracterizan por la sustitución del tejido óseo normal por tejido conectivo. En 1891 Von Recklinghausen fue el primero en describir una alteración ósea caracterizada por deformidades y cambios fibrosos que denominó “osteítis fibrosa generalisata”1. En 1938, Lichtenstein y Jaffé2 introdujeron el término “displasia fibrosa”, y reflejaron que esta alteración podía ser monostótica o poliostótica. En 1937 McCune3 y Albright et al4 relacionaron la variante poliostótica con pubertad temprana y áreas de pigmentación cutánea (manchas café con leche), asociación que en la actualidad se conoce como síndrome de McCune-Allbright y cuya incidencia es de un 4% de los casos de displasia fibrosa.
La variante monostótica supone un 70% del total de casos y en orden de frecuencia afectan a costillas, fémur, tibia, maxilar, mandíbula, calota craneal y húmero.
La variante poliostótica supone aproximadamente un 30% de los casos y clásicamente su comienzo se describe a una edad menor que la variante monostótica. En un 50% de los casos se afecta la región craneofacial. Con mayor frecuencia, y en orden decreciente: maxilar, mandíbula, hueso frontal, hueso esfenoidal, etmoides, hueso parietal y occipital5.
Marie et al6 y Riminucci et al7 establecieron que la etiología de la displasia fibrosa radica en una mutación activadora somática de la subunidad a de la proteína G de señalización (Gs-a) en células osteoblásticas, con una distribución en mosaico. La sustitución de una cys o his por arg en posición 201 conlleva una pérdida de la actividad guanosina trifosfato de Gs-a. Esto permite la activación de la adenilato ciclasa y, por tanto, la sobreproducción de AMP-c con el consiguiente incremento en la proliferación celular y una diferenciación celular inadecuada.
Estas células osteoblásticas también producen interleucina 6 en exceso, con lo que la actividad osteoclástica está incrementada. De esta forma, se producen lesiones osteolíticas expansivas que afectan al tejido fibroso y al hueso normal adyacente8.
Histológicamente la displasia fibrosa se caracteriza por una extensa proliferación de tejido fibroso que se entremezcla de forma irregular con trabéculas de hueso inmaduro. Se han descrito tres patrones diferentes por los que estas trabéculas se pueden agrupar: “letras chinas”; “puzzle” y “C&S”. Además, la matriz osteoide contiene osteoblastos inmaduros dispersos de modo irregular. La transformación maligna en la displasia fibrosa es poco frecuente, y las tasas de malignización son de un 0,5% para la variante monostótica y del 4% para la variante poliostótica asociada al síndrome de McCune-Allbright9.
La clínica relacionada con las lesiones de displasia fibrosa va a depender en gran medida de la localización y del tamaño de la lesión. De esta forma, podemos encontrarnos con lesiones de relativo gran tamaño prácticamente asintomáticas hasta lesiones de escaso volumen que, dada su localización (generalmente la región craneofacial), pueden producir alteraciones funcionales (por ejemplo, ceguera en lesiones del conducto óptico) o estéticas importantes.
Desde un punto de vista radiográfico, las lesiones de displasia fibrosa se caracterizan por un aspecto de vidrio deslustrado debido a la mezcla de elementos óseos y fibrosos. La densidad radiográfica de la lesión dependerá de la proporción relativa de estos elementos. Así pueden adoptar un patrón esclerótico, quístico (lítico) o mixto. La variante esclerótica constituye un 35% de los casos descritos y tiende a localizarse en la base del cráneo. La variante mixta es la más frecuente (40% de los casos), y el patrón quístico es el menos frecuente.
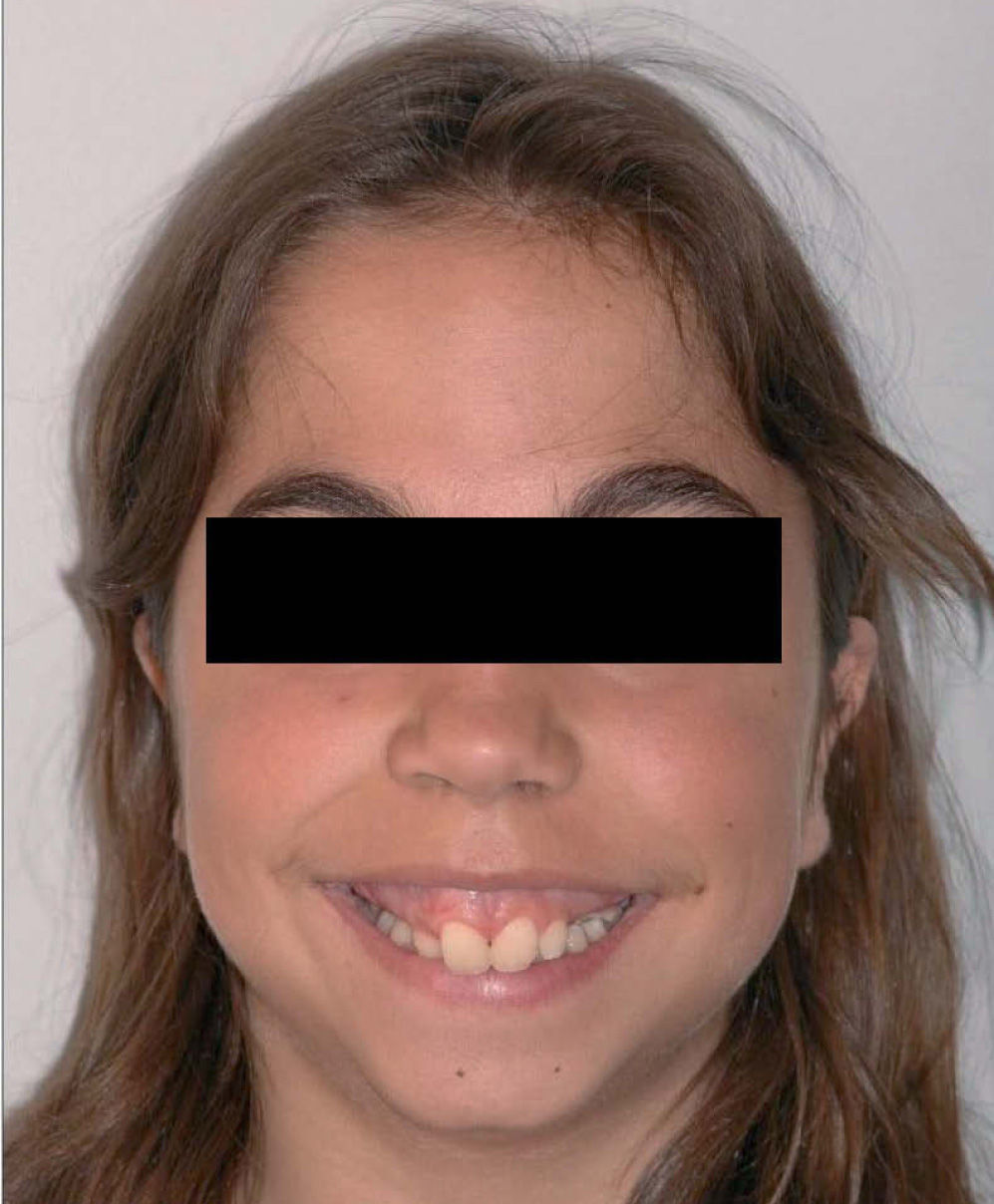
Caso clínicoMujer de 14 años que en 2007 acude a nuestra consulta remitida por su odontólogo general para valorar posible tratamiento combinado ortodóncico-cirugía maxilofacial de la deformidad dento-facio-oclusal que presentaba la paciente. En el examen clínico inicial destacaba una facies microgénica con un perfil marcadamente convexo, mandíbula hipoplásica, incompetencia labial y sonrisa gingival por excesiva altura maxilar (fig. 1). La exploración intraoral reveló la presencia de una dentición mixta de 2.a fase con ausencia de 13, 14, 23 y 24. Maloclusión de clase II con 3mm de sobremordida y 6,5mm de resalte (fig. 2).


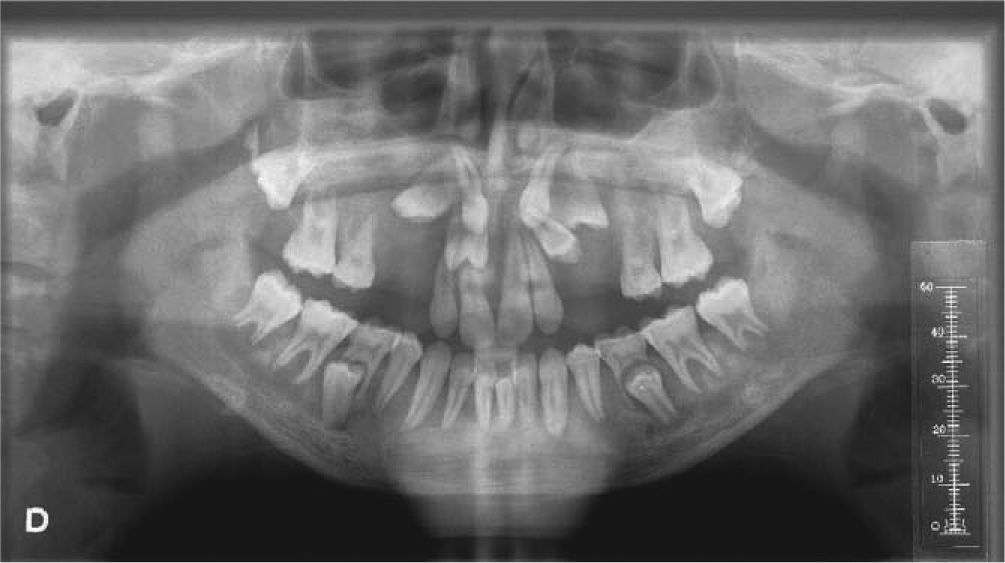
La radiografía panorámica, así como la oclusal palatina, mostraron múltiples inclusiones dentarias, así como desplazamiento hacia la línea media de 16, 26.
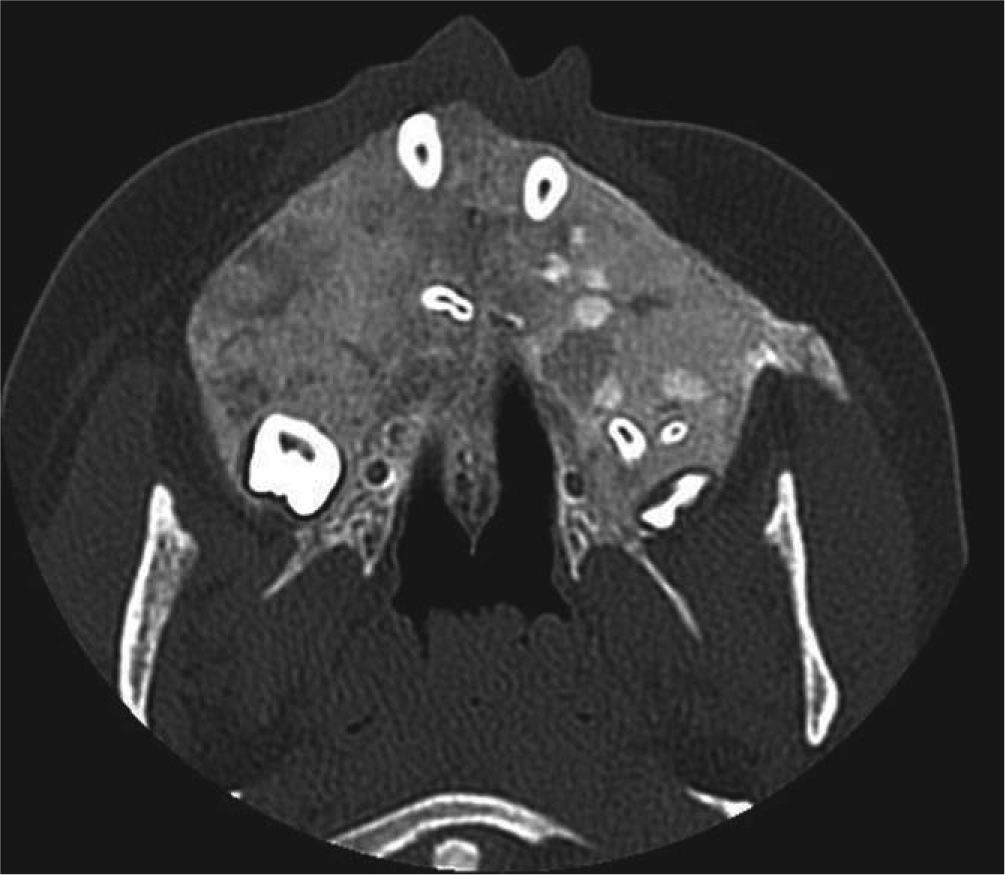
Se realizó Denta-Scan maxilar para valorar con más precisión la posición de los dientes incluidos10. Se observaron unos maxilares con aumento de densidad de forma generalizada, heterogénea, siguiendo un patrón radiológico en vidrio deslustrado. Al observar el maxilar se apreció una expansión grave de éste con ocupación parcial de los senos maxilares y retenciones dentarias múltiples con una situación muy alta y palatina de los caninos. El diagnóstico de presunción fue displasia fibrosa poliostótica (fig. 3).

Imagen axial de tomografía computarizada de maxilar superior en la que se aprecia aumento de densidad ósea de forma generalizada, heterogénea, siguiendo un patrón radiológico en vidrio deslustrado y múltiples inclusiones dentales, hallazgos compatibles con un cuadro de displasia fibrosa.
Ante estos hallazgos, entre otras medidas, se decidió realizar una biopsia, que confirmó el patrón histológico de displasia fibrosa y se profundizó en la historia clínica de la paciente que inicialmente había referido estar en tratamiento por afección renal (nefrocalcinosis) con restricción dietética de calcio.
Con 2 años de edad, la paciente había sido diagnosticada de hipercalcemia secundaria a hiperparatoridismo primario con nefrocalcinosis secundaria. Inicialmente se planteó diagnóstico diferencial con síndrome de Williams, displasia cráneometafisaria, displasia ósea (tipo Rosenberg-Lohr) o condrodisplasia metafisaria tipo Jensen. Aunque cumplía alguno de los criterios, no era concluyente para ninguna de las entidades.
La paciente mejoró con tratamiento sintomático tanto de la calciuria y de la creatinina con corticoides (prednisona 10mg/48h) + retirada de productos lácteos.
Con 3 años de edad, la paciente inicia tratamiento con pamidronato (Aredia®) 1mg/kg/2 semanas durante 5 meses. Hubo mejoría analítica de todos los parámetros de metabolismo óseo, incluida una disminución de los valores de fosfatasa alcalina. Estos datos se confirmaron con una mejoría importante en la densitometría.
Por espacio de 3 años la paciente se encontraba clínicamente muy bien, con mejoría progresiva de la densitometría y con pauta alternante de alendronato (Fosamax®).
Con 8 años de edad se apreciaron lesiones óseas metafisarias que impresionaron de displasia ósea metafisaria. Se suspendieron los bisfosfonatos durante 6 meses para valorar su reintroducción en caso necesario. Tras este período, se reintrodujeron los bisfosfonatos por un período de 2 meses para tratamiento de hipercalcemia transitoria.
Finalmente, 6 meses antes de acudir a nuestra consulta, con 13 años de edad, se reintrodujo el tratamiento con alendronato (Fosamax®) 40mg/24h. En la actualidad la paciente ha suspendido el tratamiento de forma consensuada con su nefrólogo para disminuir el riesgo de necrosis maxilar como consecuencia del tratamiento combinado ortodóncico-quirúrgico, y dados los antecedentes de administración de bisfosfonatos orales e intravenosos de larga evolución.
En la actualidad la paciente se encuentra en tratamiento ortodóncico.
DiscusiónCon la publicación de este caso clínico los autores del presente artículo pretenden exponer dos cuestiones que a su parecer son de importancia. Por un lado, el diagnóstico de una entidad infrecuente en el ámbito odontológico como la displasia fibrosa, que como se comentó anteriormente precisa en no pocas ocasiones de la integración de los datos clínicos, histológicos y radiológicos para alcanzar dicho diagnóstico. En el caso que se presenta fue especialmente complejo, porque las alteraciones óseas que se descubrieron habían sido atribuidas inicialmente a la enfermedad de base de la paciente (hiperparatoridismo primario con alteración crónica del metabolismo fosfocálcico). En este sentido, es importante para el odontólogo general conocer, si bien no en detalle, todos los aspectos de esta afección, al menos los datos básicos que le permitan orientar el diagnóstico.
En segundo lugar, se trata de un caso de displasia fibrosa que ha sido tratado de forma accidental con bisfosfonatos como parte del tratamiento de su hiperparatiroidismo primario. En términos generales, el tratamiento clásico de las lesiones de displasia fibrosa que producían sintomatología de importancia o alteraciones estéticas significativas implicaba, en la mayor parte de casos, realizar intervenciones quirúrgicas resectivas de mayor o menor amplitud en conjunción con técnicas reconstructivas, especialmente en el ámbito de la cirugía craneofacial11,12.
Por el contrario, en los últimos 10 años, se han publicado diversos estudios en los que se analizaba la efectividad del tratamiento con bisfosfonatos para la displasia fibrosa13–15. No obstante, aunque los resultados son prometedores en términos de mejora de la sintomatología y de la densitometría de las lesiones óseas, el número de pacientes incluidos en cada estudio es bajo y, además, no han sido claramente elucidados aspectos como la disminución del dolor.
En el caso que se presenta, es prácticamente imposible determinar el efecto que han podido tener los bisfosfonatos en la evolución de la displasia fibrosa sin caer en la especulación. Al no tener registros iniciales, es difícil determinar la evolución de las lesiones maxilares, por ejemplo. Lo que sí parece claro es que la paciente no ha presentado sintomatologías doloras o compresivas y, por otro lado, el aspecto de las lesiones es de lesiones calcificadas maduras aparentemente estables. Por tanto, podríamos pensar que en cierto modo los bisfosfonatos administrados a la paciente pudieran haber tenido un efecto estabilizador de las lesiones.
Para finalizar, recordar la importancia de un diagnóstico temprano de la displasia fibrosa y su derivación a especialistas (cirujanos maxilofaciales, neurocirujanos, cirujanos ortopédicos) para su valoración y tratamiento, si procede.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
