


Juvenile ossifying fibromas (JOF) are rare, locally aggressive tumours with high potentials of recurrence. The present case is that of a JOF involving the maxilla causing facial deformity, proptosis and visual impairment in a 12-year-old child which was managed successfully by surgical excision via transfacial approach, with review of the literature.
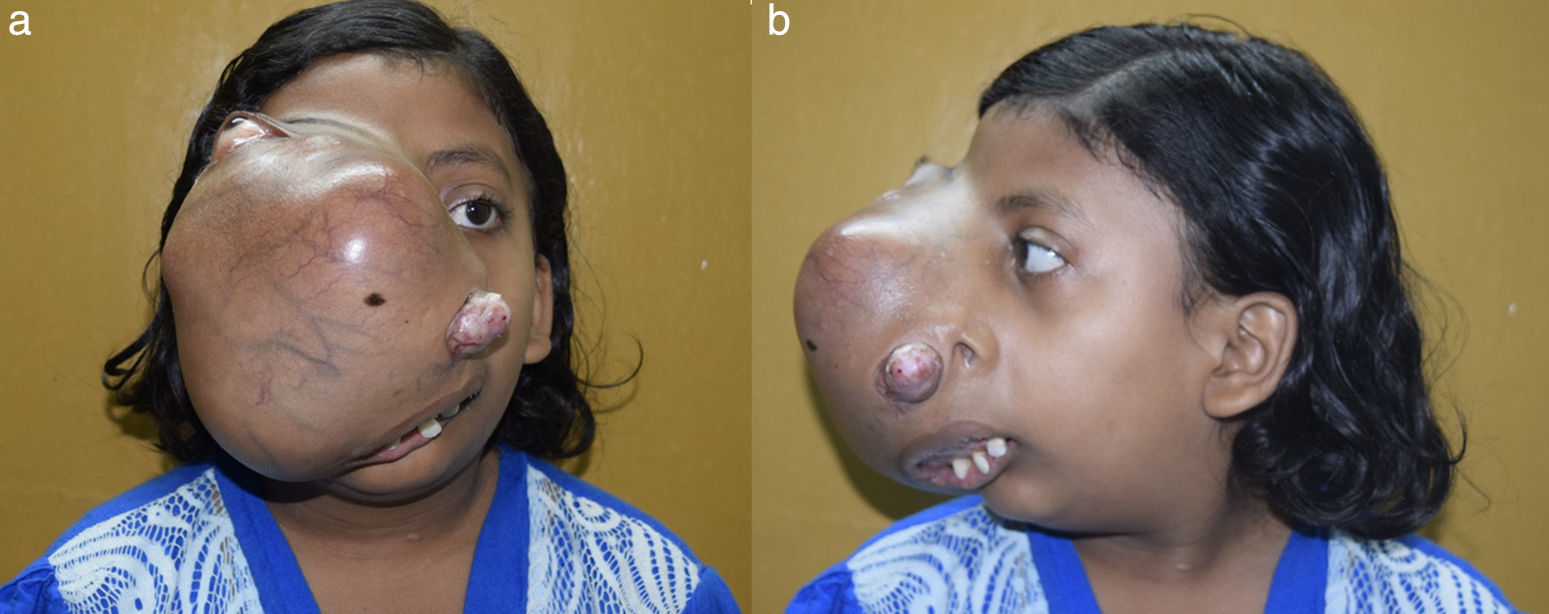
Case reportA12-year-old female child presented to our outpatient department with history of a progressive right sided facial swelling, causing facial deformity and proptosis for the past four years. The swelling pushed the right eye outwards and upwards, causing a painless, progressive diminution of vision and watering in the right eye. There was also a history of progressive nasal obstruction initially in the right nostril, which then went on to involve the left nostril (Fig. 1A and B). At presentation, the patient could only breathe through the mouth due to complete obstruction of both nostrils. She also gave a history of one episode of epistaxis which required hospital admission and conservative management. The past medical record of the child revealed that two years ago, the patient had undergone a biopsy of the mass at an outside hospital and diagnosed as having fibrous dysplasia. On examination the swelling was of the size 18cm×15cm, extending superiorly to the right eyebrow, displacing the right eye supero-laterally; inferiorly till the angle of the mandible; medially till the left side of the nasolabial fold; and laterally till the right zygomatic arch. The swelling was non-tender, with no local rise in temperature; hard in consistency. The skin over the swelling was stretched, with visible dilated veins over the swelling. The mass extended outside from the right nostril, pushing the lateral nasal wall and vestibule outwards. The mass also pushed the nasal septum to the left, causing complete obstruction to the left nostril. The patient was breathing with an open mouth, with the widening of the upper incisors. There was a widening of the right upper alveolus and obliteration of the right upper gingivobuccal sulcus by the mass. Examination of the right eye showed eccentric non-axial proptosis, visual acuity restricted to finger counting at a distance of 1m only. There was exposure keratopathy in cornea of the right eye and features of optic nerve compression.
 Front and side profile of the patient showing swelling over the right side of the face, causing distortion and displacement of right eye outwards and laterally.")
A provisional diagnosis of fibro-osseous lesion was made considering the history and clinical examination.
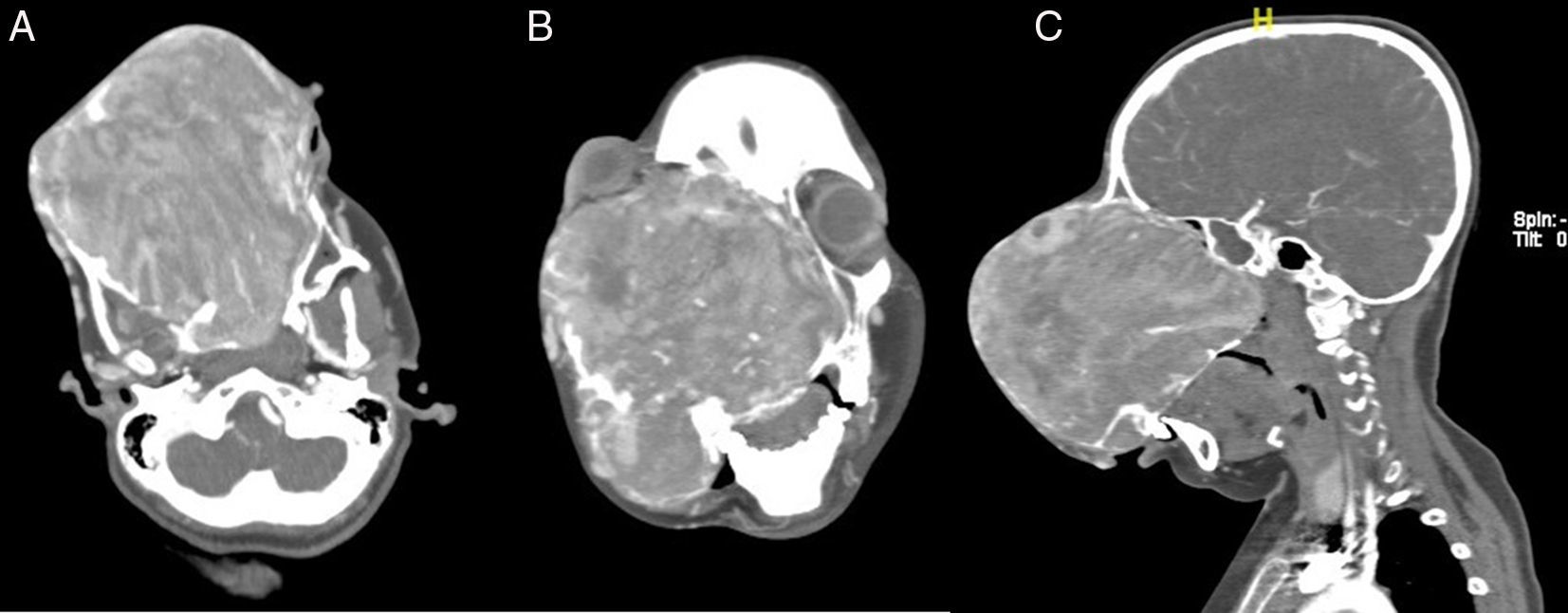
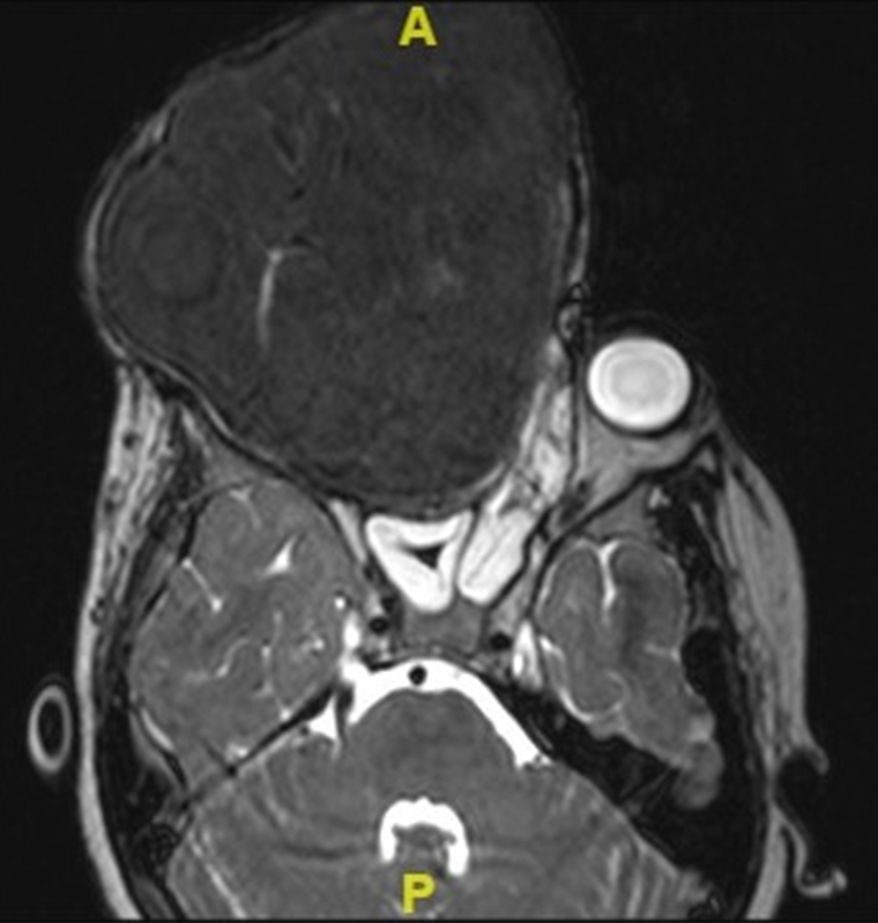
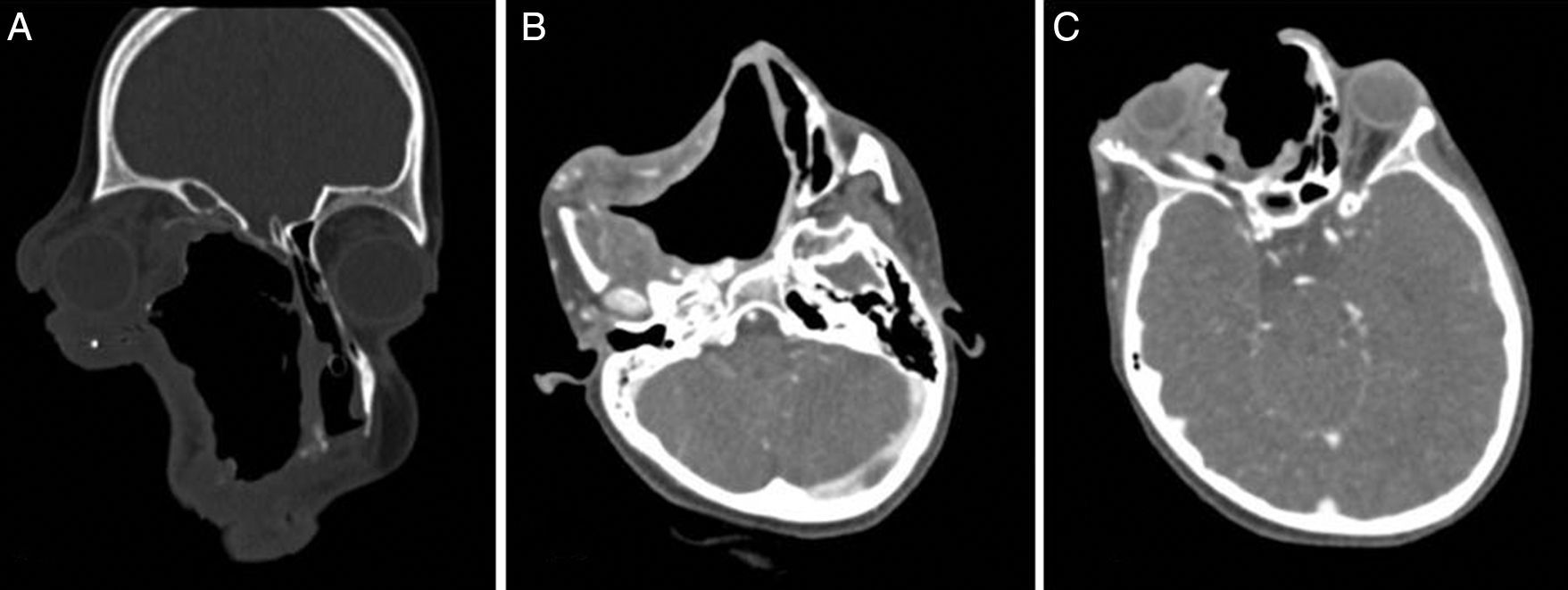
We proceeded to do a contrast enhanced 3-dimensional computed tomography scan of the skull and facial region (Fig. 2A–C). This scan showed a 17cm×12cm×11cm expansile, heterogeneously enhancing soft tissue attenuating mass lesion with multiple hyperdense areas within it; involving the right maxilla, ethmoidal bone and pterygoid plates with cortical bone thinning. No extraosseous soft tissue lesion was noted. Medially the lesion displaced the lateral wall of the right nasal cavity, causing its obliteration. Posteriorly it involved the medial and lateral pterygoid plates; superiorly extending till the inferior orbital rim, causing gross superolateral proptosis and stretching of the right optic nerve. There was no intracranial extension noted, which was confirmed by doing an MRI scan (Fig. 3).
 (Axial), (B) (Coronal) and (C) (Sagittal) sections of contrast enhanced CT scan of the nose and paranasal sinuses showing a 17cm×12cm×11cm expansile, heterogeneously enhancing soft tissue attenuating mass lesion, causing gross deformity of the face.")

Biopsy of the swelling was taken from the mass protruding from the right nostril under local anaesthesia, which confirmed the diagnosis of ossifying fibroma.
Considering the large size of the mass, the patient underwent a digital subtraction angiography (DSA) with embolization of the feeding vessels one day prior to planned excision. Right internal maxillary, right facial, right ophthalmic and left internal maxillary arteries were feeders to the tumour. Embolization of the right and left internal maxillary artery and the right facial artery was then carried out using absorbable gelatin. The child tolerated the procedure well and was asymptomatic after the embolization.
The patient was posted for a total Maxillectomy under general anaesthesia. Orotracheal intubation was achieved. Access to the right external carotid artery was obtained by making an incision in the neck, to prevent any in advent bleed. Maxillectomy was done by a standard Weber Fergusson incision, and the entire mass was excised in toto. The right pterygoid plate was drilled out as it showed a remnant of the mass. The pterygoid plates were drilled till the Vidian canal.
Grossly the tumour weighed 972.8g, which according to the best of our knowledge is the largest reported in the English literature. Histopathology of the tumour showed spindle shaped fibroblasts in a cellular stroma with strands of cellular osteoid encasing irregular osteocytes. Focal areas of multinucleated giant cells were also noted. This was suggestive of the trabecular variant of ossifying fibroma.
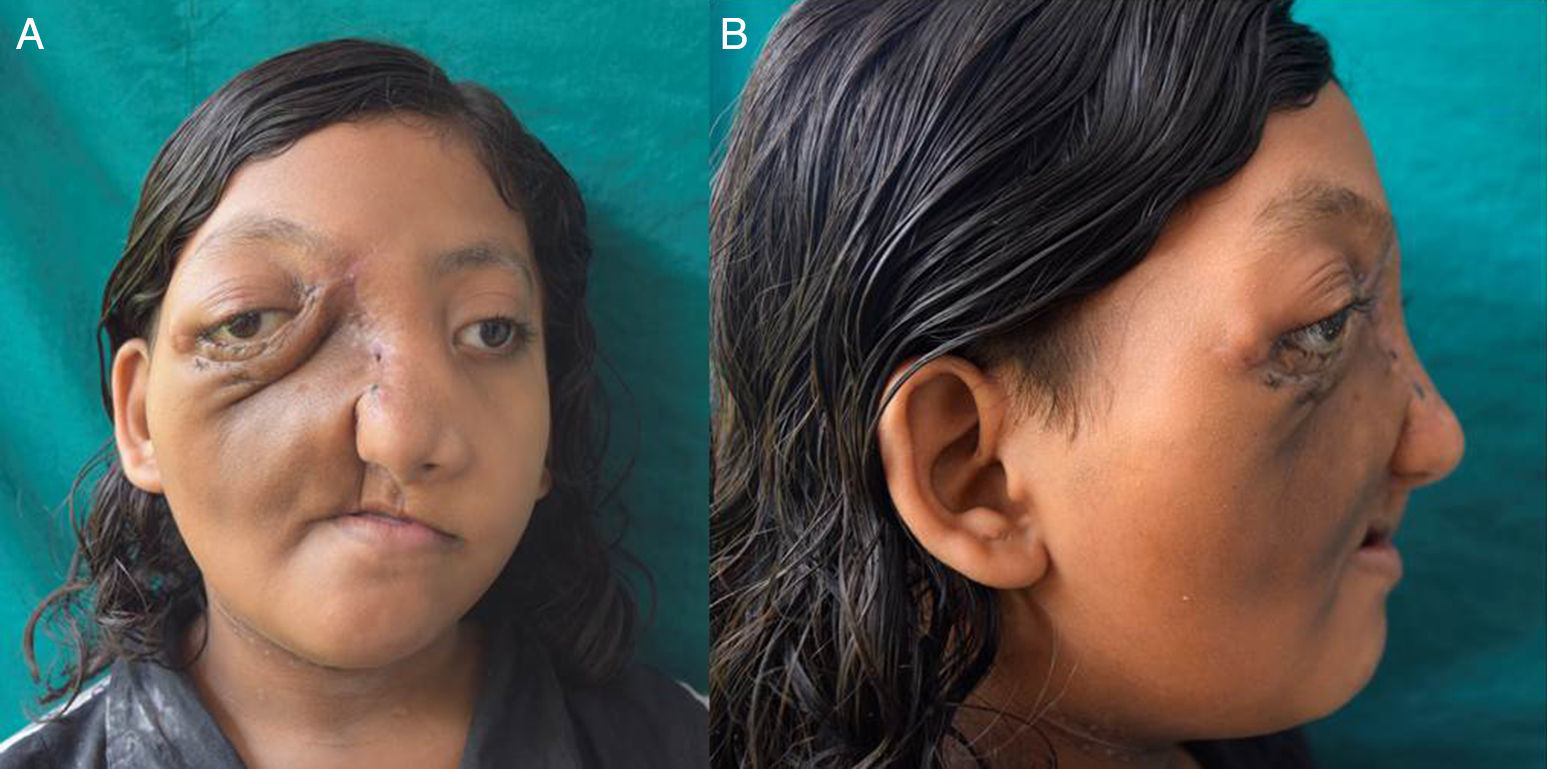
Post-operative course was uneventful. Post-operative contrast enhanced CT did not reveal any recurrence. (Figs. 4 and 5) The patient is under regular follow-up at present, and would be planned for a reconstructive surgery at a later date.
Discussion
 (Coronal), (B) and (C) (Axial) sections of contrast enhanced CT scan done post operatively.")
Fibro-osseous lesions are ones in which the bones are replaced by the formation of woven, lamellar bone with dense amorphous mineralization. These lesions are the result of maturation by cellular fibrous tissue, formed by the replaced bone.1 Amongst the odontogenic fibro-osseous lesions, juvenile ossifying fibroma (JOF) is uncommon, yet important entity. JOF is a benign neoplasm of bone with a predisposition of early age of onset, locally aggressive behaviour, typical bone pattern and high tendency to recur.2 Histologically it comprises of trabeculae of woven bone, without osteoblastic rimming and bands of cellular osteoid, within a cell-rich fibrous tissue.3 It is distinguishable from the adult type of ossifying fibroma on the basis of age, site of occurrence, microscopic appearance and clinical behaviour.
Other names of JOF include juvenile active ossifying fibroma, aggressive ossifying fibroma or young ossifying fibroma. Most common age group for its occurrence ranges from 8 to 12 years.4
JOF is thought to arise from mesenchymal cell differentiation of the periodontal ligament-which is a precursor to cementum, fibrous tissue and osteoid.1 Mutations of the tumour suppressor gene HRPT2 has been documented in these lesions by Pimenta et al.5 JOF may also be due to maldevelopment in tissues between roots of molar teeth which help in generation of bony septa, according to Lawton et al.6
85% of JOF lesions originate in the facial bones (Maxilla being more common than mandible), among which 90% arise within paranasal sinuses (ethmoid>frontal>maxillary>sphenoid sinus). 12% occur in the calvarial bones, and 4% are extra-cranial.7 Based on the bony involvement, symptoms can vary from proptosis, exophthalmos, nasal symptoms and sinusitis.8 The swelling may grow to cause gross facial deformity. The largest of the JOF we have come across in literature is of size 12cm×8cm.9 Our patient had a tumour size measuring 17cm×12cm×11cm, and weighing 972.8g, which by far is the largest that has been reported in English literature.
The differential diagnosis for JOF includes fibrous dysplasia, aneurysmal bone cyst, osteosarcoma, osteoblastoma and cement-osseous dysplasia. Its propensity to be monostotic, having rapid growth and radiologically having well-demarcated margins, differentiates it from fibrous dysplasia.9
JOF is histologically of two types, based on the age of occurrence and histologic criteria-Trabecular JOF (TrJOF) and Psammomatoid JOF (PsJOF). TrJOF is less common, with the involvement of maxilla and mandible bones; histologically there is spindle-shaped fibroblastic proliferation with a round to oval bony collections within an osteoid rim. PsJOF is more aggressive, more frequent and has a high tendency to recur. It tends to involve the orbital and sinonasal bones. Histologically it is comprised of trabeculae of immature woven bone, osteoblasts and fibroblastic spindle cell stroma in an osteoid matrix.10 Our patient is a case of a trabecular variant of ossifying fibroma.
On CT scan, eggshell thinning of the cortical bone by the mass which has a distinct boundary helps it to be radiologically differentiated from fibrous dysplasia, in which the mass blends into the surrounding bone. Ossifying fibroma on T1-weighted MRI shows intermediate signal intensity, which moderately enhances with IV contrast; and on T2-weighted MRI shows hypointense signal.11
Management of JOF is surgical. Small lesions are treated by enucleation or curettage; larger ones need an open surgical approach with radical excision. Recurrence rates vary from 30% to 58% in literature, with recurrences seen anywhere between 6 months and 7 years. Hence, a minimum five year follow up is advocated in these patients.12,13
Radiotherapy has been tried but is however contraindicated due to the risk of malignant transformation, which ranges from 0.4% to 0%. No metastasis is reported in JOF, in spite of its aggressive local behaviour.14
ConclusionOur case is different from the reported cases in literature in the following aspects:
- 1.
It is the largest JOF ever reported in literature.
- 2.
The tumour mass was causing extensive disfigurement of the face with proptosis and diminution of vision.
- 3.
The tumour had bilateral blood supply from both internal and external carotid systems.
- 4.
Histologically it was of the trabecular variant-which is rarer among the two types, less aggressive, and with a lesser tendency to recur.
The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
