


La incontinencia pigmentaria (IP2, síndrome de Block-Sulzberger) es un raro caso de genodermatosis de herencia ligado a X dominante, afectando en su mayoría a mujeres. Consiste en una serie de manifestaciones de la piel, desordenes dentarios, oculares, neurológicos, y otros. Nosotros presentamos un caso de incontinencia pigmentaria con las clásicas manifestaciones cutáneas asociado a fisura palatina.
Incontinentia pigmenti (IP2, Block-Sulzberger Syndrome) is a rare x-linked dominant genodermatosis mainly affecting females. It consists of characteristic skin manifestations and dental, ocular, neurological, and other disorders. We present a case report with classical cutaneous features diagnosed with incontinentia pigmenti associated with a cleft palate.
La incontinencia pigmentaria (IP2), también llamada síndrome de Bloch Sulzberger, es una genodermatosis rara descrita por Garrod y definida por Bloch-Sulzberger, Siemens y Bardach durante la década de 19201,2. Es un desorden neuroectodérmico que afecta piel, dientes, ojos y al sistema nervioso1–3. Se trata de una anomalía congénita ligado al X dominante (Xq28)4. El nombre de IP2 describe la característica histológica de la incontinencia del pigmento melánico de los melanocitos de la capa basal de la epidermis y su consecuente presencia en la dermis superficial que es la etapa final de esta dermatosis1.
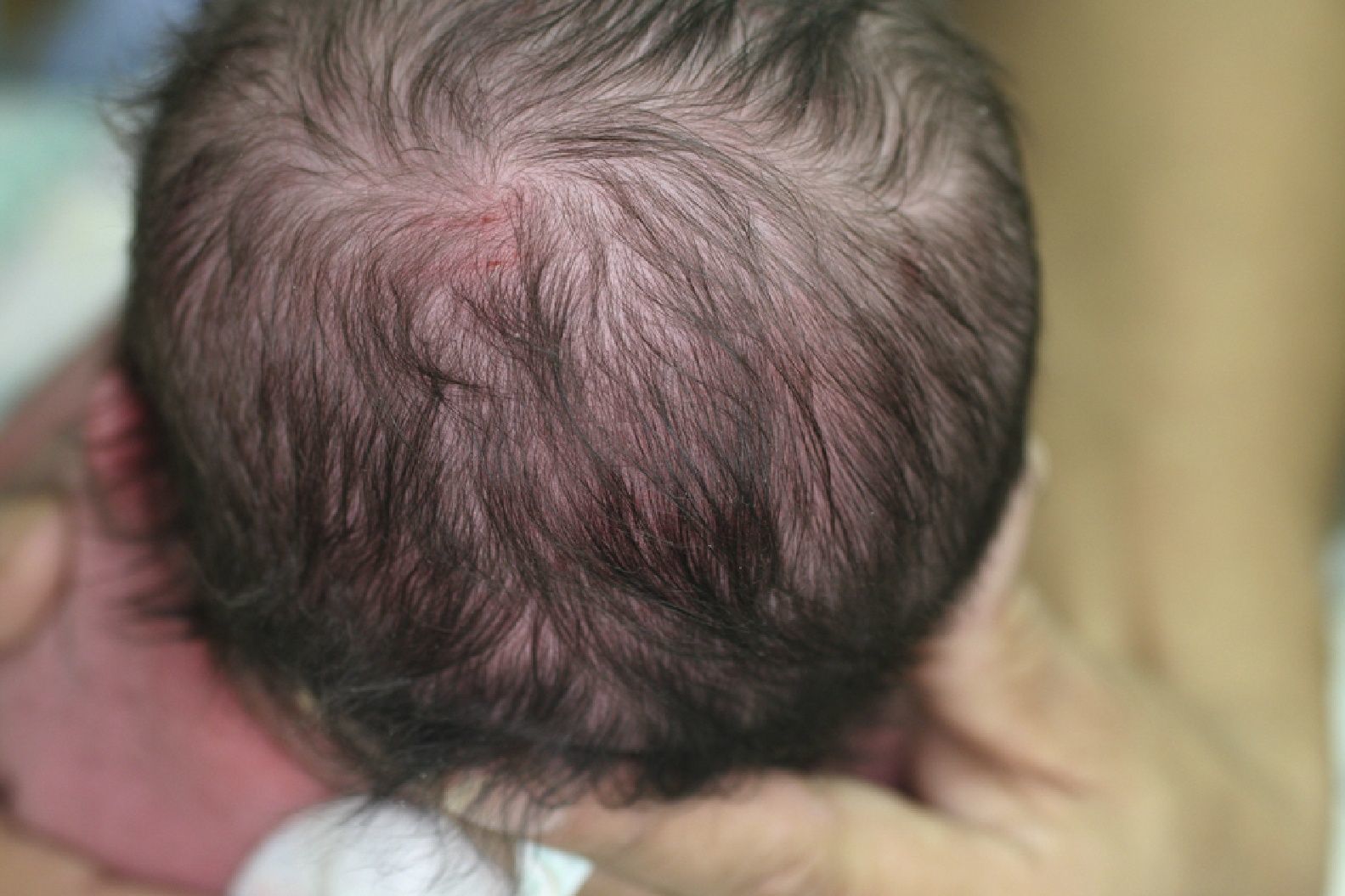
Las manifestaciones clínicas de la IP2 son variados incluso en miembros de la misma familia5. Los hallazgos en la piel generalmente son los primeros en manifestarse5,6. La manifestación más frecuente es la alopecia del vértex que ocurre en el 38 a 50% de los pacientes3,7. En 7 a 40% de los pacientes las uñas están afectadas. Estas manifestaciones cutáneas pueden verse cambiadas a lo largo de la vida, incluso las lesiones hiperpigmentadas pueden desaparecer8.
Las anormalidades cutáneas se presentan en cuatro etapas clínicas distintas. La primera se presenta al momento del nacimiento y se caracteriza por vesículas en línea o agrupadas en las extremidades del tronco y cara. Al término del primer mes pueden desaparecer, recurrir o ser reemplazadas por pápulas irregulares y lesiones inflamatorias. La segunda etapa se caracteriza por lesiones hiperqueratósicas alrededor del mes de vida. La tercera etapa usualmente entre el tercer y sexto mes se presenta con maculas café- grisáceas en patrón reticular. La cuarta etapa o «burn out» aparecen lesiones despigmentadas, pálidas y en ausencia de pelo9.
Las manifestaciones oculares, cuando están presentes, son de las más severas y generalmente están asociadas con el daño neurológico, el 35% de los pacientes presentará uno o más problemas oftalmológicos siendo el estrabismo lo más común10. Manifestaciones en el sistema nervioso central incluyen retraso mental, microcefalia, episodios convulsivos y atrofia cerebral, entre otros3,7,11.
Las manifestaciones dentales son las más frecuentes después de las dermatológicas. El 90% de los pacientes las presenta12,13. Estas manifestaciones persisten durante toda la vida, a diferencia de las cutáneas. Ambas denticiones están afectadas. La presentación más frecuente es hipodoncia, le siguen los dientes cónicos y en clavija, erupción retardada y otras alteraciones menos frecuentes, tales como dientes impactados o fusiones. Se han informado casos de IP con labio y paladar figurado como una de las manifestaciones menores de la enfermedad14,15.
Este caso muestra un paciente con fisura palatina (CP) asociado a un síndrome autosómico dominante como es el caso de IP2.
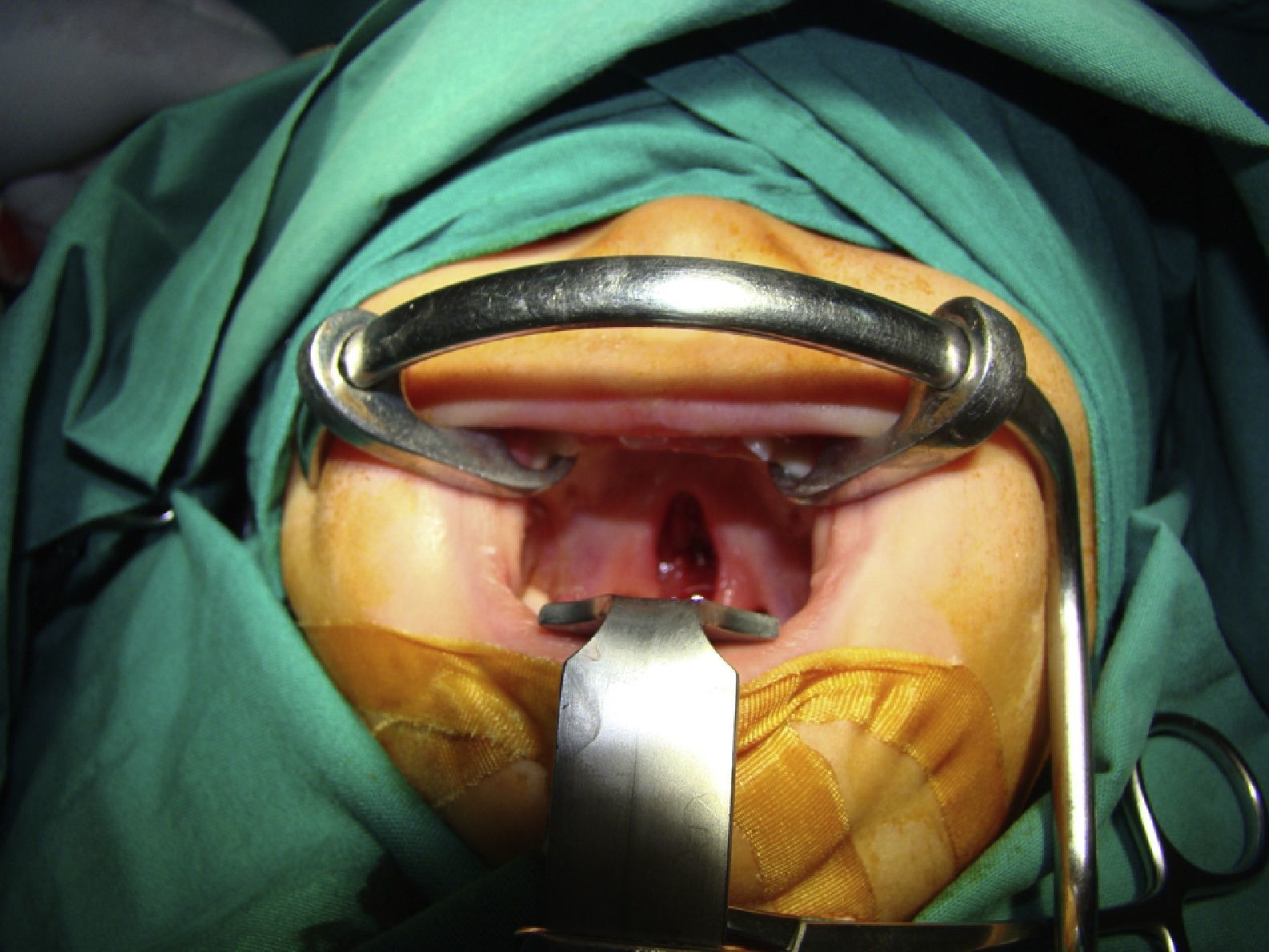
Reporte del casoPaciente femenino de 1 año 2 meses de edad vista en el Hospital de Carabineros Santiago, Chile, con parto en madre de 28 años por cesárea. Con un peso de 3.163 gramos, talla 49cm y perímetro craneal cercano a 35cm. Al momento del nacimiento se observan lesiones vesiculosas en extremidades inferiores, zona glútea y axilas que se rompen al retirar el unto de la piel quedando lesiones eritematosas y descamativas. Se le diagnosticó en primera instancia epidermolisis bulosa, la cual posteriormente fue descartada. Destacaba además zona de alopecia en vértex y paladar fisurado (figs. 1–3).



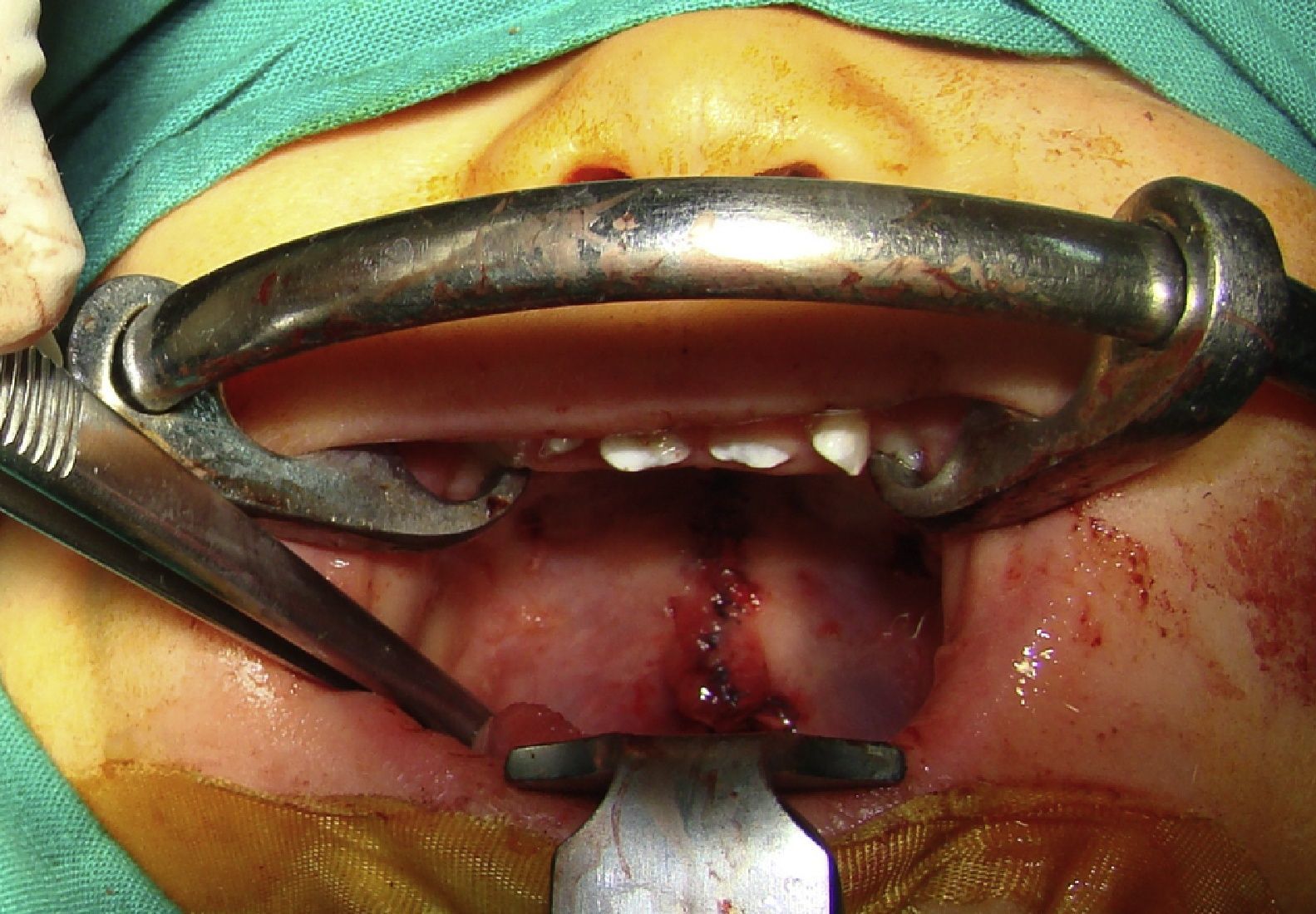
Se da refiere a oftalmología que realiza examen de fondo de ojo observándose epitelio pigmentario con poco pigmento en forma generalizada, resto normal. Evaluación por neurólogo normal. Dermatólogo plantea el diagnóstico de IP Se realiza biopsia de piel encontrándose la muestra dentro de los limites histológicos normales, pero se recomienda otra nueva muestra. Visto por genetista clínicamente se confirma el cuadro de herencia ligada a X dominante, ya que la madre presentó las mismas lesiones al nacimiento. Se derivó también a la unidad de maxilofacial por CP. A los 10 meses de edad fue operada de paladar fisurado, reconstituyendo funcionalmente los anillos musculares del velo palatino y evolucionó sin presentar complicaciones postoperatorias.
DiscusiónLa incontinencia pigmentaria está considera como una poli-displasia mixta ectodérmica y mesodérmica. Donde las mujeres son afectadas en un rango de 37:1, siendo más común en caucásicos, en la revisión de Carney de 653 casos, 593 eran mujeres y 16 varones (3). Al menos 900 casos se han descrito en la literatura, pero la prevalencia de la IP2 es desconocida7,9.
Carney en su publicación del año 1976 encontró que el 79,8% de los pacientes tenían alteraciones en el pelo, ojos, dientes, sistema nervioso central y otras estructuras anormales3. En la tabla 1 se exponen los criterios propuestos para el diagnóstico de la IP2.
La IP2 tiene anormalidades características en la dentición que deben tomarse en cuenta una vez que empiecen a erupcionar los dientes. Hipodoncia, retardo de la erupción y formas cónicas coronarias, ya sea de la dentición primaria o secundaria, pueden parecer. Estos cambios son similares a aquellos observados en la forma de la displasia ectodérmica hipohidrótica, algunos autores sugieren una relación entre ésta y la IP26. Por lo tanto se debe estar al tanto con controles periódicos realizando un adecuado examen bucal por la posible aparición de alguna anormalidad que comprometa la salud oral.
En los resultados del estudio de Carney3 el 1,1% (5 casos) presentaron fisura labial o anormalidades en el paladar, como paladar ojival, fisura labio palatina, hipoplasia palatina y fisura labial parcial. En el año 1976 Brett15 publicó un caso que incluía fisura labio palatina lo cual le presento dificultades para alimentarse e intubación por algunas semanas. Samman reportó en el año 1959 un caso de IP2 con fisura parcial de labio entre otras alteraciones cutáneas, dentales y hemiplejia12. Yell a su vez aportó con un caso único de fisura labio palatina bilateral13. En una publicación de 40 casos de IP2 se encontró que dos hermanas y su madre tenían el paladar fisurado aislado14. Por lo tanto, casos de CP son difíciles de encontrar en la literatura.
Según las estadísticas de Jone‘s et al, el porcentaje de fisuras asociados a síndromes fue 13,8% en FLP (de un total de 574 pacientes), 41,8% en CP aislada (de un total de 328 pacientes) y 78,3% (de un total de 46 pacientes) en insuficiencia velo faríngea, para lo cual consideró como síndrome el hecho que el niño tuviera asociado a la fisura dos o más malformaciones mayores o tres o más malformaciones menores no explicadas por antecedentes familiares. Mostrando con sus resultados, al igual que en nuestro caso, que los síndromes están más asociados a CP aislada16.
Nuestra paciente presentó las características cutáneas típicas de la IP2 propias de la etapa 1, sumándose a esta los signos del cabello y las alteraciones oftalmológicas. De gran importancia fue la historia clínica de la madre para llegar a una hipótesis diagnóstica confirmada posteriormente con el genetista. Se debe mencionar que la lactante fue operada de la fisura a los 10 meses de edad debido a cuadros de enfermedad respiratorias que imposibilitaron las fechas óptimas protocolizadas para el cierre palatino según nuestro protocolo quirúrgico de fisuras palatinas.
La cirugía de reconstrucción del velo se realizó sin infortunios, encontrándose buena calidad de tejido muscular para lograr la reconstrucción de los anillos musculares, recuperando la funcionalidad del esfínter velo faríngeo en su totalidad. Por lo tanto, esta manifestación de la enfermedad se puede llevar a cabo sin consideraciones especiales a la hora de la programación quirúrgica.
Aunque la IP2 sea considerada una condición escasa, se debe tener en cuenta al tener casos de anormalidades bucales y dentarias en infantes que presenten otra manifestación mayor asociada al cuadro dentario. El papel del cirujano dentista, en estos casos, es preponderante. Éstos deberán estar atentos a los hallazgos clínicos y radiográficos de la condición para poder distinguirla de otras formas de displasias ectodérmicas. Además estar pendiente de que alteraciones que se presentan al nacimiento pueden anteceder a otras en el área estomatológica, estando así preparados para etapas de prevención y tratamiento de estas alteraciones.
Algunos autores responsabilizan al azar la relación que tiene la IP2 con las CLP14 y se llega a la pregunta si realmente existe una analogía entre el síndrome y la patología de CP.
