


Introducción: Los tumores neuroectodérmicos primitivos (PNET, de primitive neuroectodermal tumors) son una familia de neoplasias malignas de células pequeñas y redondas, que derivan de la cresta neural. Se distinguen tres tipos: PNET del sistema nervioso central, PNET del sistema nervioso autónomo y PNET periféricos. Los más frecuentes dentro del grupo de PNET periféricos son el neuroepitelioma periférico y el sarcoma de Ewing, que se consideran la misma neoplasia pero con diferente grado de diferenciación. Casos clínicos: Presentamos dos casos de PNET periféricos, uno de aparición en la región cervical y otro originado en el cóndilo mandibular.
Discusión: Los PNET son neoplasias muy raras y altamente agresivas. En todos ellos aparecen células redondas pequeñas poco diferenciadas y una traslocación cromosómica característica del gen EWS. En general se considera que tienen un pronóstico desfavorable. Además, la baja frecuencia de estos tumores, así como la escasez de casos publicados hacen difícil valorar el tratamiento más adecuado.
Introduction: Peripheral primitive neuroectodermal tumors (PNET) are a family of smallround cell tumors of presumed neuroectodermal origin. This broad family can be subdivided into three major groups: PNET from the central nervous system, PNET from the autonomic nervous system or peripheral PNET. Ewing's sarcoma and peripheral neuroepitelioma, the two most frequently encountered members of the peripheral PNET family, are considered to represent a spectrum according to the extent of neuroectodermal differentiation, ranging from the least differentiated (Ewing's sarcoma) to the most differentiated (peripheral neuroepithelioma).
Case report: We present a patient with a peripheral neuroectodermal tumor located in the neck and another one with a peripheral neuroectodermal tumor of the mandibular condyle. Discussion: Peripheral neuroectodermal tumors are a very rare and aggressive tumors. They characteristically reveal the presence of small round cells and a translocation of the gene EWS. The prognosis in overall is very poor. Due to the small numbers of cases published the best treatment is not well defined.
Los tumores neuroectodérmicos primitivos (PNET, de primitive neuroectodermal tumors) son una familia de neoplasias malignas de células redondas y pequeñas, que derivan de la cresta neural. Pueden aparecer en cualquier grupo de edad y no se han observado diferencias en su incidencia en virtud del sexo1.
De acuerdo con la bibliografía2-4, se distinguen tres tipos de PNET:
- 1.
PNET centrales, que abarcan tumores que surgen del sistema nervioso central, como el meduloblastoma.
- 2.
Neuroblastoma, incluyendo los tumores que surgen del sistema nervioso autónomo.
- 3.
PNET periféricos, que son los que surgen fuera del sistema nervioso central o autónomo.
La clasificación y terminología de este tercer grupo es algo complicada y controvertida3-5. El grupo está constituido por: sarcoma de Ewing (óseo y extra-óseo), neuroepitelioma periférico del hueso y tejidos blandos, tumor de Askin (que se trata de un neuroepitelioma periférico que se localiza en la región torácica y pulmonar), tumor neuroectodérmico melanótico (o melanoma maligno), ectomesenquimoma y meduloepitelioma periférico.
Los tumores más frecuentes dentro del subgrupo de PNET periféricos son el neuroepitelioma periférico y el sarcoma de Ewing.
En este artículo presentamos el primer caso que se ha descrito de PNET en el cóndilo mandibular y un segundo caso también excepcional de PNET en la región cervical, además de una revisión de la literatura.
Caso 1: tumor cervicalVarón de 29 años sin antecedentes personales ni familiares de interés, que acude a nuestra consulta para valorar una masa latero-cervical izquierda, de crecimiento progresivo y varios meses de evolución, que le produce disfonía, tos seca y disnea. Niega pérdida de peso, astenia o anorexia, y cuenta sudoración profusa nocturna, así como cefalea holocraneal frecuente. A la exploración se palpa una masa dura, adherida a planos profundos en el tercio inferior cervical izquierdo, quese extiende hacia la línea media, sin palparse adenopatías locorregionales.
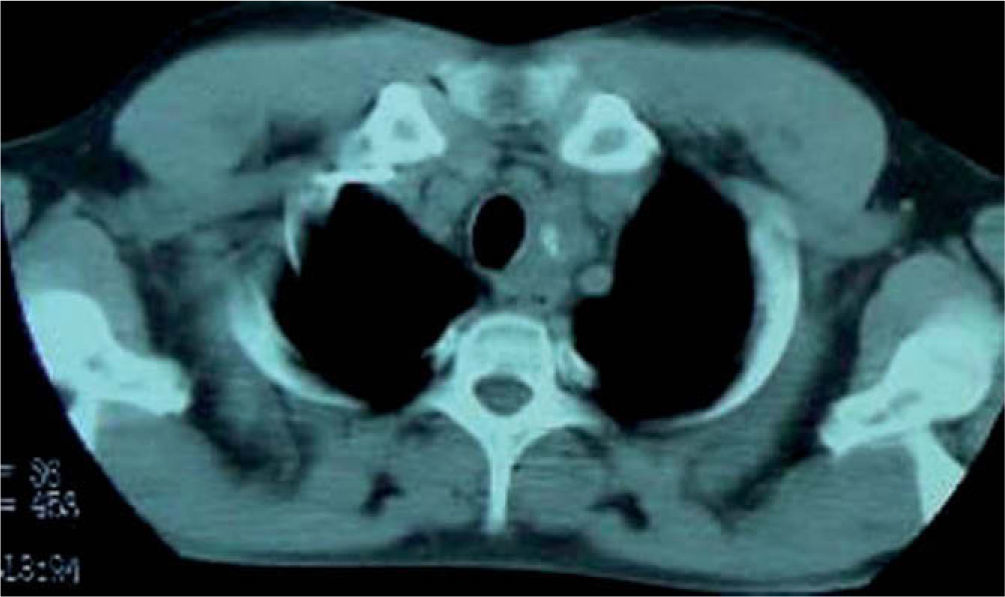
La tomografía axial computarizada (TAC) revela una tumoración de 7 x 2 cm, desde el lóbulo tiroideo izquierdo hasta el cayado aórtico, que produce cierta estenosis traqueal y desplazamiento del esófago (fig. 1).

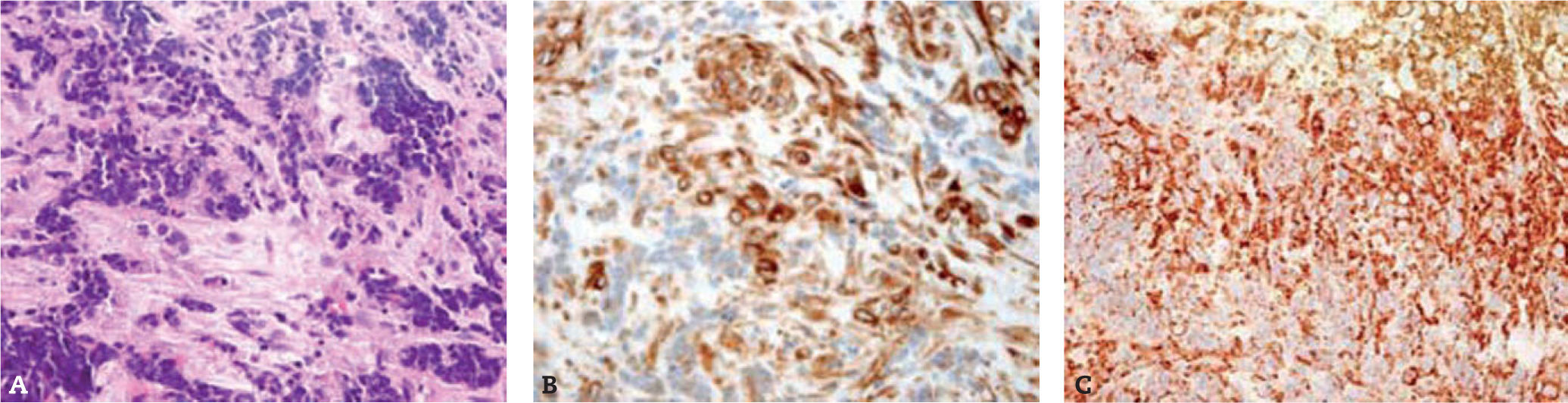
Los cortes histológicos muestran un tejido conjuntivo infiltrado en ciertas zonas por una neoplasia que se dispone formando grandes nidos de células de tamaño pequeño o intermedio (fig. 2).

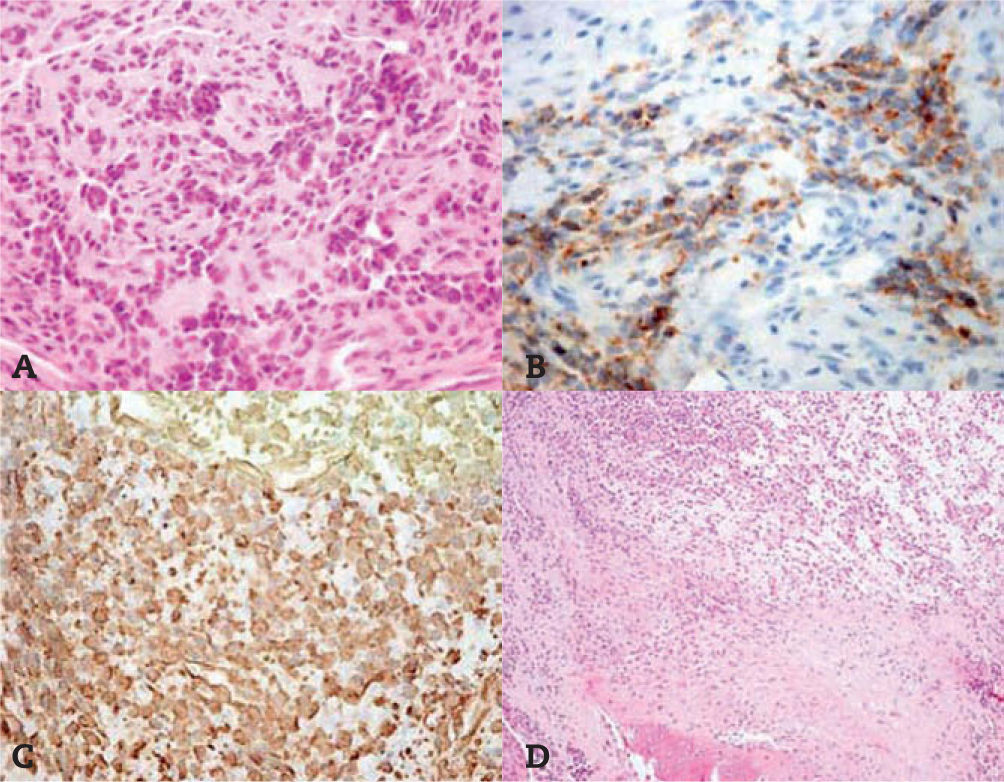
A. La tinción con hematoxilina-eosina muestra un tejido conjuntivo infiltrado por nidos de células pequeñas y redondas, atípicas, de núcleos hipercromáticos y escaso citoplasma. B. Se observa de forma muy focal y aislada la positividad para vimentina. C. Todas las células son positivas para CD99.
En el estudio inmunohistoquímico las células tumorales son reactivas exclusivamente para CD99, enolasa neuroespecífica y, débilmente, para sinaptofisina. La positividad nuclear para el anticuerpo de proliferación Ki-67 oscila entre el 25 y el 50% de los núcleos de las células tumorales.
Con el diagnóstico obtenido mediante la biopsia, se programa al paciente para recibir tratamiento quimiorradioterápico. Recibió 4 ciclos de quimioterapia, seguidos de tratamiento radioterápico con una dosis total administrada de 55,8 Gy, tras el cual presentó esofagitis y epitelitis de grado 2. Tras la radioterapia, el paciente recibió 6 ciclos de poliquimioterapia.
Los días siguientes al inicio del tratamiento presentó una mejoría progresiva de su estridor respiratorio, que desapareció por completo, así como una disminución del tamaño de la tumoración latero-cervical mayor de un 50%. Tras finalizar todos los ciclos de tratamiento se objetivó remisión completa de la enfermedad.
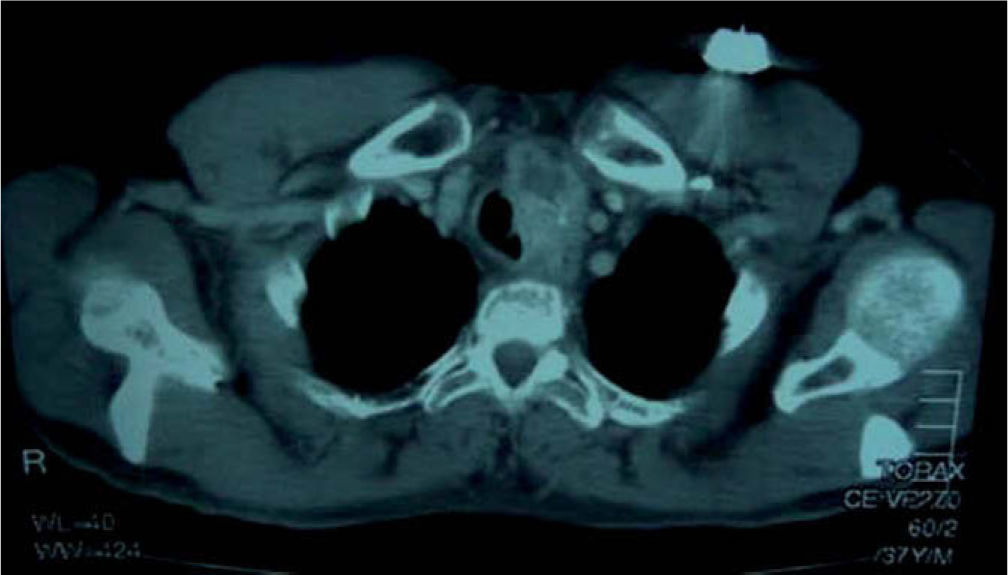
El paciente permaneció asintomático durante 7 años hasta que acude a la consulta refiriendo disnea y tos de un mes de evolución y en la TAC se objetiva recidiva mediastínica con extensión pulmonar (fig. 3). Se realiza traqueostomía por compresión traqueal del tumor y se inicia tratamiento citotóxico con 4 ciclos de poliquimioterapia. Presentó una supervivencia de un año hasta su fallecimiento por causa tumoral.

Recidiva mediastínica tumoral. Se visualiza a nivel cervical bajo, proyectándose hacia el tórax, mediastino superior y paratraqueal izquierdo, una tumoración de partes blandas homogénea, irregular, con calcificaciones en su interior, con algunos nódulos y proyecciones intratraqueales.
Mujer de 45 años, sin antecedentes personales ni familiares de interés, que nota un chasquido mandibular izquierdo en relación con la apertura mandibular y una tumoración palpable preauricular dolorosa que no mejoró con tratamiento con antiinflamatorios no esteroideos, por lo que acude a Urgencias de Cirugía Maxilofacial.
A la exploración física presenta asimetría facial, y a la palpación se aprecia un aumento de consistencia en región preauricular izquierda, arco cigomático y ángulo mandibular izquierdo. Este aumento de consistencia se halla mal delimitado, sin poderse definir clara tumoración.
En la cavidad oral se aprecia induración irregular del espacio masticador izquierdo, sospechosa de afectación tumoral. La paciente presentaba una apertura oral de 40 mm. No presentaba adenopatías cervicales ni supraclaviculares palpables.
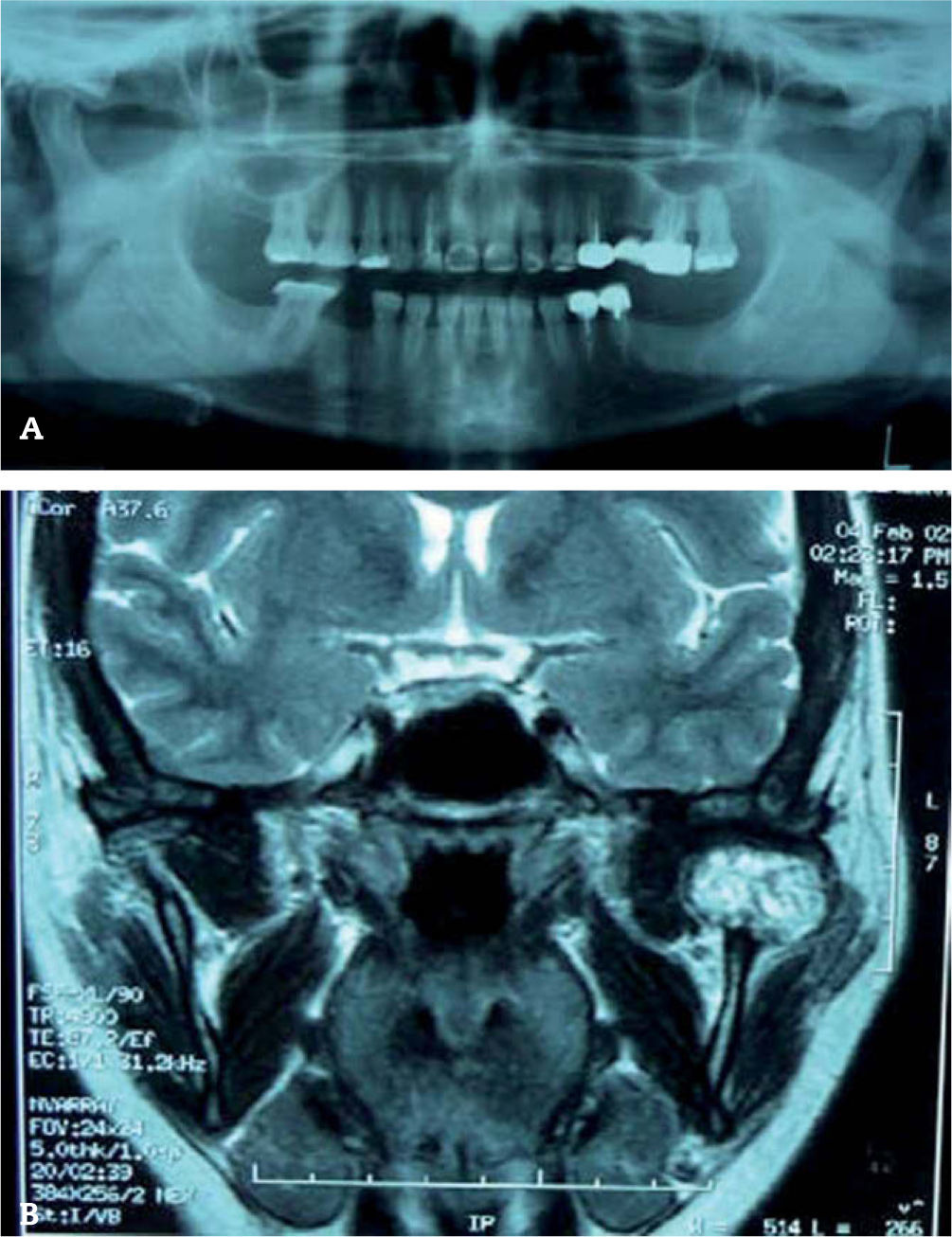
En las pruebas de imagen aparece una tumoración de carácter sólido en hemimandíbula izquierda, de 2,5 x 2 x 1,8 cm en la región del cóndilo mandibular, de bordes bien delimitados, que sugiere una lesión agresiva, de probable origen óseo, objetivándose destrucción de rama mandibular izquierda de carácter lítico (fig. 4).
 se visualiza lesión nodular de aproximadamente 2,5 cm, centrada en hueso, con destrucción del mismo.")
Se realizó punción-aspiración con aguja fina con diagnóstico anatomopatológico de tumor de células redondas, negativo para citoqueratinas, antígeno leucocitario común, sinaptofisina y CD99.
Finalmente se confirmó el diagnóstico de PNET con una biopsia tumoral en la que se mandaron 5 fragmentos irregulares de tejido de color rojizo que se diagnosticaron como "tumoración de células redondas y pequeñas infiltrando hueso".
Se realizó un estudio inmunohistoquímico sobre el material fijado en formol incluido en parafina en el que tan solo se observó positividad con vimentina y CD99 (fig. 5). El índice de proliferación medido como positividad nuclear con MIB-1 era de aproximadamente el 15% de la celularidad tumoral. Dado el escaso material examinado, no fue posible determinar si la tumoración correspondía a un tumor de Ewing o a un neuroepitelioma periférico. El análisis citogenético no fue realizado.

A y D. Tumoración de células PAS positivas pequeñas y redondas, de citoplasma escaso, de tamaño pequeño, núcleo redondeado de cromatina fina, sin nucleolo, que se disponen formando nidos o bien formando un patrón "en sábana". B. Positividad para CD99. C. Células positivas para vimentina.
La TAC y la gammagrafía ósea no mostraron la presencia de enfermedad metastásica.
Se decidió comenzar tratamiento quimioterápico y valorar tratamiento quirúrgico/radioterápico según la respuesta. Se dieron 5 ciclos durante 4 meses. Tras el tercer ciclo precisó ingreso por neutropenia febril.
La resonancia magnética nuclear (RMN) tras el tratamiento quimioterápico sugería la posible progresión del tumor, por lo que se decidió dar tratamiento radioterápico planificado con intención radical, durante un mes y medio. La irradiación se hizo en dos fases, con una dosis total sobre tumor y margen reducido de 60 Gy. Después del tratamiento radioterápico pre-sentó mucositis grado 2, otitis externa y epitelitis grado 2 en el campo de tratamiento.
En la TAC de control se objetivaron hallazgos compatibles con restos/recidiva del PNET de la paciente, por lo que se decidió tratar con otros 5 ciclos más de quimioterapia, siguiendo el mismo esquema anterior, durante otros 4 meses.
Actualmente han pasado 6 años y la paciente continúa libre de enfermedad, en tratamiento de rehabilitación dental.
DiscusiónLos tumores neuroectodérmicos periféricos son neoplasias sumamente raras que forman parte de una familia de tumores con un origen neuroectodérmico común. En todos ellos aparecen células redondas pequeñas poco diferenciadas, inmunológicamente positivas para "013" (un anticuerpo monoclonal frente al gen p30/32 MIC2), y una translocación cromosómica característica del gen EWS. Muchos de ellos presentan características clinicopatológicas parecidas, siendo necesario estudios inmunohistoquímicos, ultraestructurales y citogenéticos para alcanzar un diagnóstico correcto2, 4.
Los PNET periféricos pueden aparecer en cualquier parte del cuerpo, pero se originan normalmente en la pared torá-cica (tumor de Askin), el tronco, el abdomen o la pelvis. Los PNET del aparato genital femenino son muy infrecuentes6. Se dan también en la cabeza y el cuello, aunque de forma excepcional. Actualmente hay muy pocos casos publicados en la literatura científica de PNET de origen mandibular1, 3-5, 7. Se han descrito en región mentoniana y parasinfisaria. En la literatura no hay casos reportados en localización de cóndilo mandibular y son muy escasos los reportes de PNET en localización cervical3, 8.
La clínica de presentación suele ser una masa dolorosa de crecimiento progresivo. En nuestros pacientes aparece también una masa preauricular que produce dolor y desplaza la musculatura masticatoria en el segundo caso presentado, y como una masa latero-cervical de crecimiento progresivo, con la consiguiente estenosis traqueal (disnea, disfonía, tos seca) y desplazamiento esofágico, en el primer caso presentado. Votta et al1 publicaron el caso de una paciente que presentaba un tumor neuroectodérmico en la sínfisis mandibular, que aumentó notablemente de tamaño durante el embarazo, con recesión del crecimiento en el postparto.
Las pruebas de imagen en los PNET nos permiten evaluar el tamaño y la extensión del tumor, estudiar la resecabilidad y detectar metástasis a distancia. Además, podemos analizar la estructura interna tumoral, lo cual nos puede ayudar a elegir la estrategia terapéutica más adecuada y predecir la posible respuesta al tratamiento9. Dick et al8 estudiaron la utilidad de la TAC y de la RMN en el diagnóstico y el seguimiento de un amplio grupo de pacientes con PNET y vieron que ambas se complementan ya que determinan la extensión tumoral del tejido blando con igual eficacia, pero mientras que en la TAC se ve mejor la afectación ósea y permite encontrar pequeñas metástasis pulmonares, la RMN es muy útil para detectar invasión de la pared torácica. Además
observaron que los tumores tendían a desplazar estructuras más que a encapsularlas. La ecografía de nuestro caso de PNET condíleo mandibular muestra una masa de ecogenicidad heterogénea de carácter sólido y bordes bien delimitados. Los hallazgos con ultrasonidos en los pocos casos publicados describen la lesión como hipoecoica, pudiendo presentar áreas quísticas anecoicas que corresponden a zonas de necrosis9.
En concordancia con nuestros pacientes, otros casos de PNET publicados presentan normalmente en la TAC y en la RMN un reemplazamiento de la señal normal con aumento en T2, disminución en T1 y captación postcontraste intravenoso de forma heterogénea8, 9. Los tumores de gran tamaño (aproximadamente 5 cm de diámetro) contienen algunas áreas quísticas o de necrosis, y raramente están calcificados.
A pesar de todo ello, tanto la clínica como las imágenes del PNET son inespecíficas y el estudio histológico del tumor es fundamental para confirmar el diagnóstico y diferenciarlo de otros tumores. Histológicamente lo más característico es hallar células redondas y pequeñas que en ocasiones se disponen en nidos, cordones o lóbulos, aunque el 10-20% son células fusiformes10. En algunos casos publicados se han identificado células rabdoides y plasmocitoides [1] . Es frecuente encontrar rosetas que suelen ser del tipo Homer-Wright, que indican diferenciación neuronal. Sin embargo, en algunos casos de neuroepitelioma periférico son escasas y pasan desapercibidas.
Inmunohistoquímicamente, la positividad para MIC2 (CD99) es muy útil porque se identifica en la mayoría de los casos en el neuroepitelioma periférico y en el sarcoma de Ewing. Sin embargo, la presencia de CD99 no es específica de los PNET; el linfoma linfoblástico, los linfomas de Hodgkin y no Hodgkin y el rabdomiosarcoma alveolar y embrionario pueden presentar positividad para el CD992 También son útiles otros marcadores neurológicos, como la enolasa neuronal específica, la sinaptofisina, la cromogranina, los neurofilamentos y la proteína S-100. Con la microscopia electrónica se suelen observar células pequeñas y redondas con escaso citoplasma y grandes núcleos ovoides. Se pueden ver las interdigitaciones y las elongaciones celulares que contienen gránulos neurosecretores, en ocasiones asociados a microtúbulos y neurofilamentos.
Los estudios citogenéticos y moleculares aportan una información adicional y definitiva porque la típica translocación cromosómica t(11;22) (q24; 12) que ocurre entre los genes EWS y FLI-1 se ha identificado en el 85-90% de los neuroepiteliomas periféricos y sarcomas de Ewing1-7,9,11. También se puede dar la t(21;22)(q2;q12). Actualmente se ha descubierto una tercera translocación -t(7;22)-, que se ha relacionado con un peor pronóstico, al desarrollar una enfermedad más agresiva2.
Gran parte de los autores consideran al sarcoma de Ewing como la misma entidad que el neuroepitelioma periférico, dado que comparten la translocación t(11;22) y expresan elevados niveles de glucoproteínas p30-32 (un producto del gen MIC2 único en estos dos cánceres). Sin embargo, otros autores los separan, puesto que el sarcoma de Ewing no presenta gránulos neurosecretores, siendo considerado una forma más primitiva e indiferenciada2, 12. En general, se considera que ambos constituyen el mismo tipo de tumor con diferentes grados de diferenciación, desde el más diferenciado (neuroepitelioma periférico), que presenta abundantes rosetas de Homer-Wright o de Flexner-Wintersteiner en su histología, además de células con citoplasma eosinófilo con abundante cromatina y nucleolos prominentes, inmunoreactividad para dos o más marcadores neurales o evidencia de diferenciación neural y gránulos neurosecretores en el microscopio electrónico, hasta el menos diferenciado (el sarcoma de Ewing), cuyas células muestran un citoplasma claro y escaso, con una cromatina muy fina y grisácea y nucleolos muy discretos, además también contienen glucógeno y tienen pocas rosetas de Homer-Wright1. La tinción positiva con múltiples marcadores neurales es también una característica del sarcoma de Ewing.
El diagnóstico diferencial de los tumores de células pequeñas y redondas en la cabeza y el cuello incluye: osteosarcoma, neuroblastoma, linfoma, metástasis de carcinoma de células pequeñas del pulmón, rabdomiosarcoma, tumor desmoplásico de células pequeñas redondas, melanoma y sarcoma de Ewing extraóseo1, 6.
En cuanto al pronóstico de los PNET periféricos, en general se considera que es bastante desfavorable, con una supervivencia libre de enfermedad de aproximadamente el 50% en 3 años y del 30 al 45% en 5 años3. Sin embargo, el uso de múltiples agentes quimioterápicos de forma combinada, el tratamiento quirúrgico y el tratamiento radioterápico han mejorado el pronóstico en la última década. El lugar de origen del PNET parece ser un factor pronóstico importante. Los que se originan en la cabeza y el cuello, así como en el tórax, tienen un pronóstico intermedio comparado con los tumores paraespinales o de la región escapular, que se asocian a un pronóstico favorable, y los tumores abdominales, los cuales no responden al tratamiento. Por otra parte, los PNET orbitarios se han relacionado con un comportamiento menos agresivo.
La presencia de metástasis a distancia en el momento del diagnóstico oscila entre el 14 y el 50% e implica una elevada mortalidad13. Existe una tendencia general para desarrollar de forma temprana metástasis a distancia, especialmente hacia el pulmón, el hígado y el hueso. Normalmente se dan por vía sanguínea, aunque también puede participar la diseminación linfática7.
La baja frecuencia de estos tumores, así como la escasez de casos publicados, hacen difícil extrapolar datos que ayuden a conocer el pronóstico o el tipo de tratamiento más adecuado. El protocolo de tratamiento de estos tumores altamente agresivos incluye multiquimioterapia, cirugía y en algunos casos radioterapia. La radiación se recomienda en función del tamaño y el lugar de origen del tumor, la histología, la edad del paciente y la extensión de la enfermedad antes y después del tratamiento quirúrgico. Tras el tratamiento es fundamental el seguimiento del paciente durante años, ya que la recidiva o la aparición de metástasis a distancia ocurre con elevada frecuencia.
