



El tumor de células de Merkel es un tumor cutáneo raro, de elevada malignidad, mal pronóstico y baja supervivencia. Se caracteriza por su tendencia a la invasión ganglionar y vascular, y por un alto porcentaje de recurrencia locorregional en el año siguiente a la extirpación quirúrgica. Afecta a adultos entre los 60 y 80 años, y se localiza preferentemente en cabeza y cuello. Presentamos el caso de un varón de 85 años que acude por masa glútea ulcerada de 4 meses de evolución. El diagnóstico se realizó por estudio histopatológico e inmunohistoquímico. Un diagnóstico precoz y un tratamiento adecuado son importantes para mejorar el pronóstico de estos enfermos.
Merkel cell tumour is a rare skin tumour of high malignancy, poor prognosis and low survival. It is characterized by its tendency to lymph node and vascular invasion and by a high percentage of locoregional recurrence in the year following surgical removal. It affects adults between 60 and 80 years of age and often occurs in the head and neck. We present the case of an 85-year-old man presenting with an ulcerated gluteal mass of 4 months’ evolution. Diagnosis was by histopathological and immunohistochemical study. Early diagnosis and appropriate treatment are important to improve the prognosis of these patients.
El carcinoma de células de Merkel es un tumor cutáneo raro y de elevada malignidad. Está constituido por células pequeñas con características endocrinas y epiteliales. Afecta a adultos de ambos sexos entre los 60 y 80 años. Se localiza preferentemente en áreas fotoexpuestas de cabeza y cuello con intenso daño solar. Clínicamente se presenta como un nódulo cutáneo o subcutáneo eritemato-violáceo, no doloroso y de rápido crecimiento. El diagnóstico se basa en la clínica, histopatología e inmunohistoquímica. Debido a la baja frecuencia y la edad avanzada de los pacientes, no se dispone de estudios prospectivos y no existe un claro algoritmo en el tratamiento.
Presentamos un caso de tumor de Merkel de localización y presentación atípicas como una gran masa ulcerada localizada en glúteo.
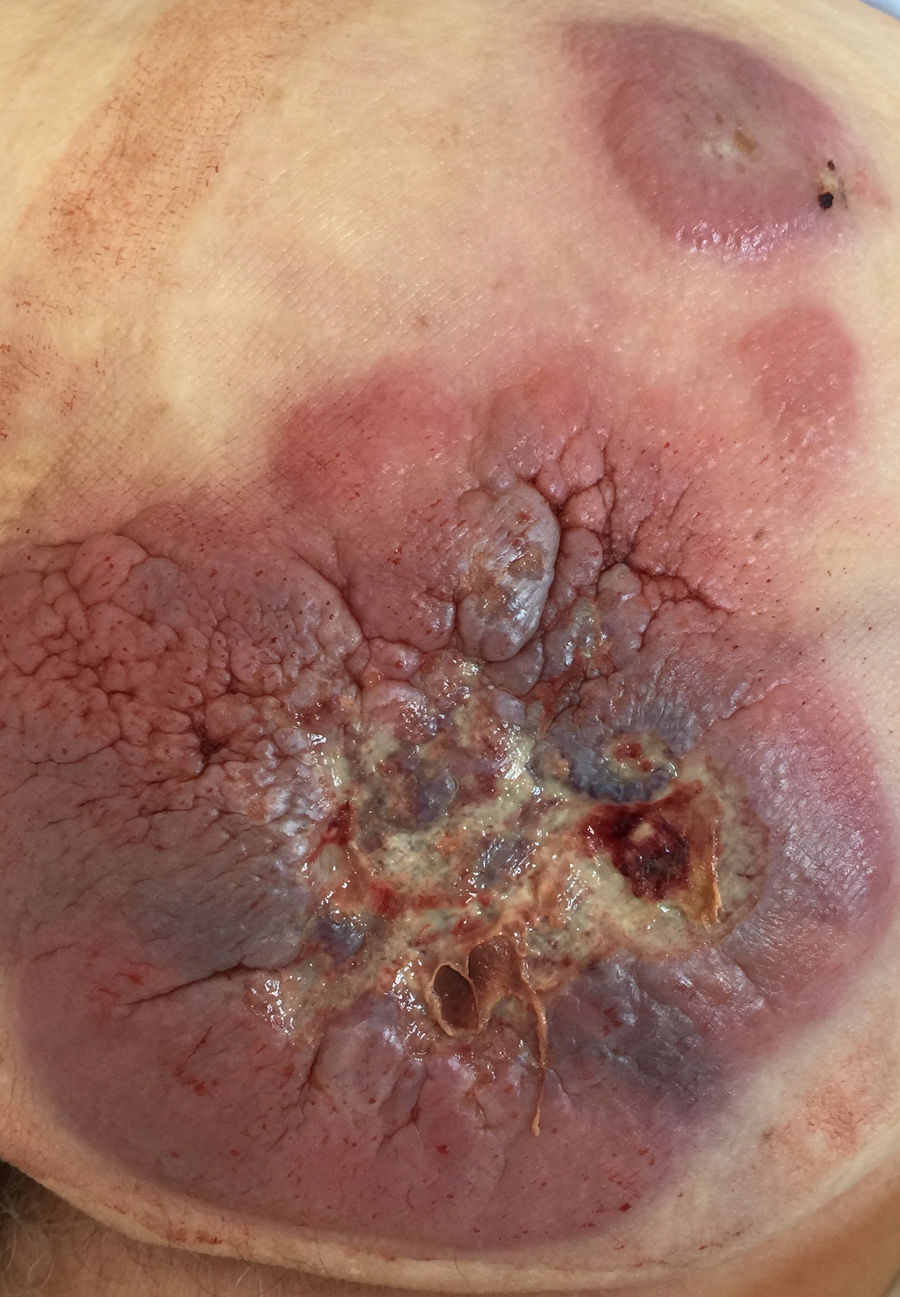
Caso clínicoVarón de 85 años con antecedentes de cardiopatía isquémica, carcinoma vesical de bajo grado y varios tumores basocelulares extirpados que acude por sangrado glúteo. A la exploración presentaba una masa de 15×15cm indurada, adherida a planos profundos y ulcerada junto con lesiones satélites en nalga izquierda (fig. 1) y adenopatías de tamaño patológico en ambas ingles.
Resultados
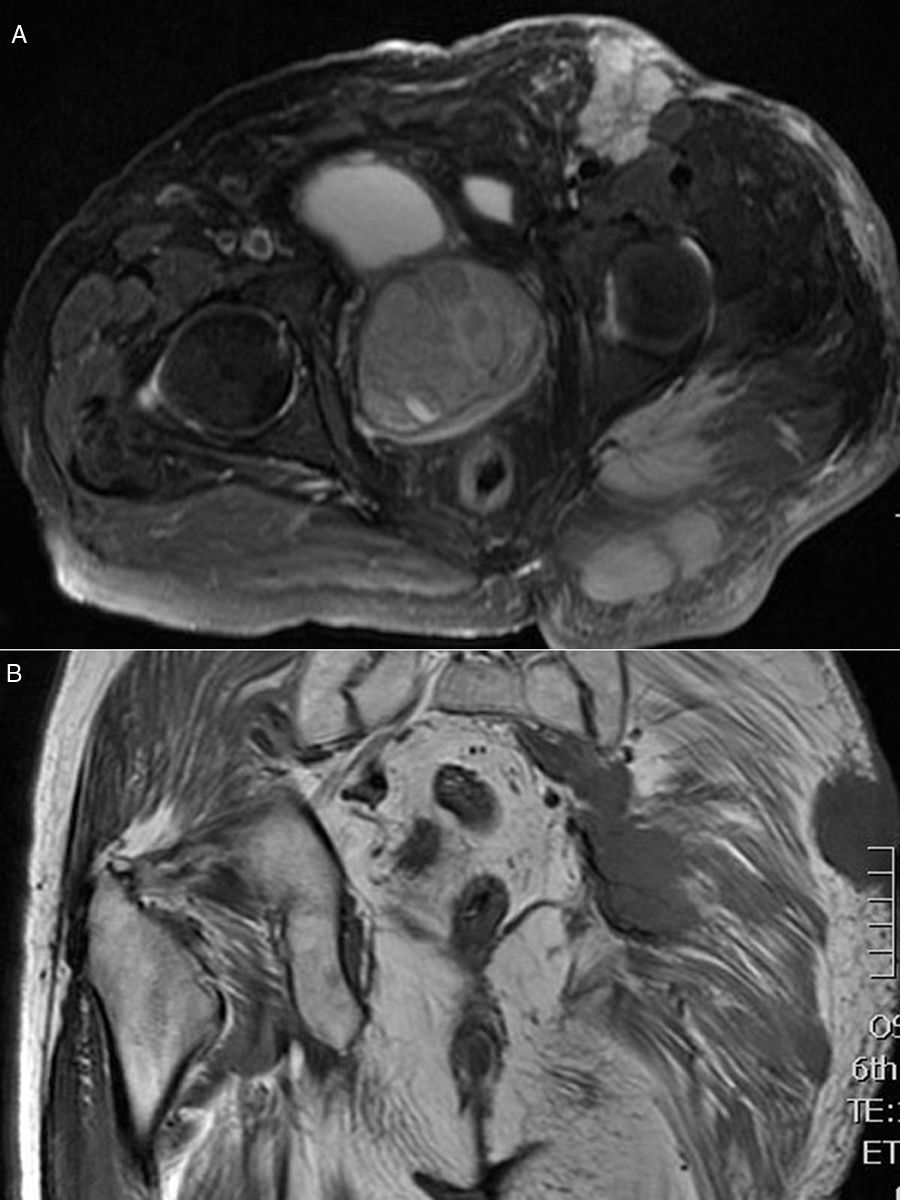
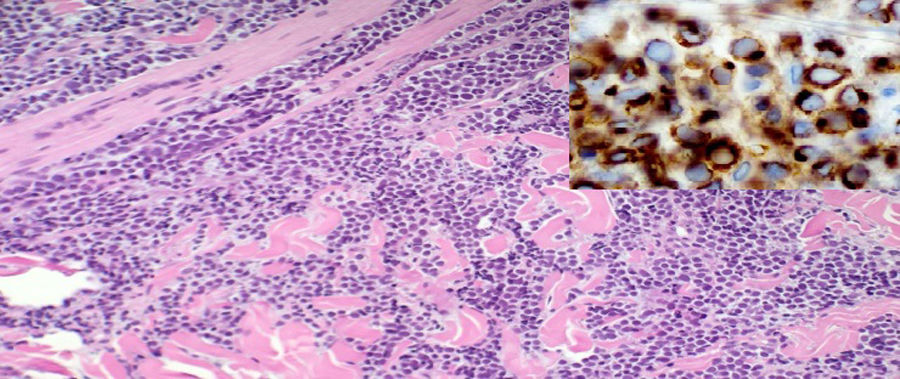
Se realizaron la tomografía axial computarizada (TAC) tóraco-abdominal y la resonancia magnética nuclear (RMN), que mostraron voluminosas masas de partes blandas en región glútea izquierda, visualizando al menos 3 lesiones tumorales en el espesor del tejido celular subcutáneo, una en la musculatura glútea y otra en región inguinal izquierda, junto con una masa-bloque adenopático en región retroperitoneal izquierda con extensión a cadena ilíaca común y externa ipsilaterales (figs. 2A y B). Se realizó biopsia incisional. La anatomía patológica fue informada como carcinoma neuroendocrino cutáneo primario, carcinoma de células de Merkel (CCM) (fig. 3).
Discusión Corte axial de la RMN. B) Corte coronal de la RMN.")
.")
El CCM es un tumor neuroendocrino cutáneo primario, infrecuente, de mal pronóstico y baja supervivencia. Se caracteriza por ser proclive a la invasión ganglionar y vascular, asociadas ambas a un alto porcentaje de recurrencia locorregional dentro del primer año de la extirpación del tumor1.
Se presenta habitualmente en adultos de raza blanca, mayores de 65 años, aunque existen casos publicados en pacientes jóvenes portadores del síndrome de displasia ectodérmica congénita2. Preferentemente afecta a áreas fotoexpuestas: 55% en cabeza y cuello, 40% en miembros y 5% en tronco2–4. En casi un tercio de los casos este tumor se asocia con otras neoplasias cutáneas como la enfermedad de Bowen, el carcinoma basocelular o el carcinoma espinocelular, como en nuestro paciente4. Estudios más recientes implican al poliomavirus en la carcinogénesis del carcinoma de Merkel5. Puede aparecer como una neoplasia secundaria en pacientes con alteraciones inmunológicas de distintas etiologías: leucemia linfática crónica, linfoma de células B, mieloma, VIH y también en pacientes que han recibido trasplante de órganos o en tratamiento prolongado con inmunosupresores6,7. En estos pacientes la agresividad y la mortalidad es aún mayor. El compromiso inmunológico es un factor de riesgo para el CCM.
La lesión cutánea tiene aspecto aplanado (placa o pápula) o sobreelevado (nódulo), es de color rojo-violáceo y su crecimiento es rápido. La afectación epidérmica es rara, por lo que pocas veces se ulcera.
El diagnóstico anatomopatológico es difícil ya que puede ser fácilmente confundido con metástasis cutáneas de otros tumores de células redondas y pequeñas: sarcoma de Ewing, tumor de células pequeñas de pulmón (oat cell) o neuroblastoma. Para el diagnóstico de certeza se requiere el empleo de inmunohistoquímica positiva a CK20 en patrón de punto paranuclear8.
Las metástasis a distancia establecen la estadificación siendo la piel el lugar más frecuente seguido de ganglios linfáticos regionales, hígado, pulmones, hueso y cerebro. En el tratamiento se incluyen la cirugía amplia con extirpación del ganglio centinela, radioterapia, quimioterapia, pero no hay un protocolo de actuación establecido debido a su baja incidencia9,10. En nuestro paciente, dada la edad y la extensión del tumor se optó por tratamiento sintomático que llevó al fallecimiento del paciente, un mes tras el diagnóstico.
El interés de nuestro caso radica en la existencia de un tumor cutáneo infrecuente asociado a una localización y una presentación poco habitual, lo que dificulta el diagnóstico clínico inicial de la lesión.
