



The purpose of this study is to present our series of Ewing sarcoma cases and the survival data obtained in the medium term, using a multidisciplinary therapy protocol.
Material, methods and resultsForty-one Ewing sarcomas were diagnosed, treated and followed-up in our hospital between 2004 and 2009 with an average age of 18.29 years. Seventy-eight percent were to Ewing sarcoma of the bone, the femur being the most frequent location. Sixty-eight percent had a localised stage at the time of diagnosis. At the end of follow-up, 40% of the patients did not survive, most died within the first 5 years of follow-up.
DiscussionIn Spain, Ewing sarcoma is the most common primary malignant bone tumour in childhood, ahead of osteosarcoma. Its survival rate has increased greatly in the last 40 years, improvement attributable mainly to the aggressive use of chemotherapy and to multidisciplinary treatment, but its prognosis remains very poor, especially for those with metastasis at diagnosis, the main adverse prognostic factor. Because of its high mortality, many authors consider it a disseminated disease from the beginning, with non- detectable micrometastasis that condition final survival.
ConclusionsEarly diagnosis and multidisciplinary therapy in referral centres are the best strategies currently available to us to provide these patients the maximum possibilities of cure of this disease.
El objetivo de este estudio es presentar nuestra serie de casos de sarcoma de Ewing y los datos de supervivencia obtenidos a medio plazo utilizando un protocolo de terapia multidisciplinar.
Material, método y resultadosCuarenta y un sarcomas de Ewing fueron diagnosticados, tratados y evolucionados en nuestro centro entre 2004 y 2009 con una media de edad de 18,29 años. Hasta un 78% correspondieron a Ewing óseo siendo el fémur la localización más frecuente. El 68% presentó un estadio localizado en el momento del diagnóstico. Al final del seguimiento la supervivencia no llegó al 40% de los pacientes, falleciendo la mayoría en los primeros 5 años de seguimiento.
DiscusiónEn España, el sarcoma de Ewing es el tumor óseo maligno primario más frecuente en la infancia, por delante del osteosarcoma. Su tasa de supervivencia ha aumentado mucho en los últimos 40 años, mejoría atribuible fundamentalmente al uso agresivo de la quimioterapia y al tratamiento mutidisciplinar, pero su pronóstico sigue siendo muy pobre sobre todo en aquellos que presentan metástasis al diagnóstico, principal factor pronóstico adverso. Dada su alta mortalidad, son muchos los autores que lo consideran como una enfermedad diseminada desde el principio, con micrometástasis no detectables que son las que condicionan la supervivencia final.
ConclusionesEl diagnóstico precoz y el tratamiento multidisciplinar en centros de referencia son las mejores estrategias con las que contamos en la actualidad para proporcionarles a los pacientes las máximas posibilidades de curación de esta enfermedad.
Ewing sarcoma is a primitive primary malignant tumour of bone described in 1921 by James Ewing, who called it diffuse endothelioma of bone.1–3 Its exact histogenesis is unknown, it appears to derive from the bone marrow cells,4 corresponding to a poorly differentiated form of the primitive neuroectodermal tumour.2 It consists of small, blue, round malignant cells with a common chromosomal translocation, in almost cases, the EWSR1 gene in chromosome 22 and a member of the family of ETS transcription factors in chromosome 9.1,3,5
Ewing is the second most common primary malignant bone tumour in childhood, ahead of osteosarcoma,1,6 and the most common in Spain, according to the national paediatric cancer registry of 2016.6 Eighty percent appears in people under the age of 20, it is rare in children under 5 and people over the age of 30,1 it is more common in males and extremely rare in black people. Its approximate annual incidence is 3 cases per million white people and people under the age of 21.1,4,7,8
Its most common site is in the diaphysis or metaphysodiaphyseal segments of the long bones, the femur being the site in 20–27%,3,8 followed by the tibia, fibula and humerus. Unlike osteosarcoma, it frequently appears in the flat bones of the axial skeleton, essentially the pelvis, thoracic wall (in this case it is called Askin's tumour) and the sacrum.1,3 Up to 20% are extraskeletal sarcomas, soft tissue tumours with no bone involvement but with the same histology as the Ewing bone tumour, mainly sited in the paravertebral region, thoracic wall and lower limbs.1,9 They are rarely sited in the nervous system, either as a primary or metastatic, these are usually epidural extraskeletal Ewing tumours.
A painful, palpable tumour (Fig. 1) together with redness and heat is the most common form of presentation. Up to a fifth also have constitutional symptoms due to the release of cytokines10 such as fever, asthenia, loss of appetite and weight loss,1,3 that can even lead to an erroneous diagnosis of osteomyelitis.4 It most commonly presents at stage IIB of the American Joint Committee on Cancer (AJCC)4 and around 20% have metastasis on diagnosis, the lung being the most common site, but it is estimated that many more present with micro-metastases on diagnosis which the current diagnostic techniques cannot are unable to detect. These would be responsible for the poor prognosis of Ewing tumours that are classed as “localised”.3,10,11

The tumour appears radiologically as an osteolytic lesion with a moth-eaten or permeative pattern, periosteal “onion skin” reaction and a large soft tissue non-mineralised soft tissues. In the long bones, the mixed lythic/blastic pattern with an area of sclerosis predominates.1,4,9,12
Treatment of Ewing sarcoma is multidisciplinary; chemotherapy is the basis and surgical resection the fundamental pillar for its local control. Although traditionally surgery and radiotherapy were at the same level in terms of local control, it is now known that surgery provides better results for disease-free survival than radiotherapy alone,3,4,10 and also avoids the risk of secondary radio-induced sarcomas.4 Thus, in patients with no evidence of metastatic disease at time of diagnosis, the protocols for the treatment of Ewing sarcoma include pre- and post-operative polychemotherapy (it has been known since the nineteen seventies that chemotherapy improves survival and the combination of several drugs is better than monotherapy),1surgical resection and radiotherapy if there are affected margins or recurrence.1,4 Isolated radiotherapy is reserved for large and unresectable axial lesions in order to avoid mutilating surgery3 and for patients with lung metastases who have responded well to chemotherapy, even when they have achieved full remission, since the rate of lung recurrence can be reduced by up to 50% with radiotherapy.1 Intensive chemotherapy and megatherapy with autologous haematopoietic stem cell transplantation are good alternatives for treating patients at risk.1,6 Innovations in the treatment of this tumour include new chemotherapy agents, in particular gemcitabin-docetaxel, trabectedine, anti-IGF1R antibodies, fenretinide, sirolimus and deforolimus.3
Survival rates have increased from 10–15% to 65–70% over the past 40 years,3 essentially due to the aggressive use of chemotherapy and multidisciplinary treatment.4,6 However, the prognosis remains poor, especially for patients with disseminated disease or early recurrence;1,3,4,6 disease-free survival at 5 years being 9–30%.10 The most important prognostic factors are the presence of metastasis on diagnosis, which is the principal adverse prognostic factor, the site and the size of the tumour, tumour load and the biological response to neoadjuvant chemotherapy, and the time from diagnosis to recurrence.1,3 Other factors at the time of diagnosis such as being over the age of 14, elevated LDH levels and previous symptoms of short duration also appear to be associated with a poor prognosis for the disease.6
Material and methodWe undertook a retrospective analysis, approved by the Institutional Review Board, between 2004 and 2009 (72 months) of the Ewing sarcomas diagnosed in our hospital, a referral centre for musculoskeletal tumours for the autonomous regions of Andalusia, the Canary Islands and Extremadura. To obtain a follow-up period of 6 years, we set the end of follow-up as December 2015. During this time, 45 Ewing sarcomas in 45 different patients were diagnosed by means of a combined search performed between the Pathological Anatomy Department and the Musculoskeletal Tumour Unit of the Orthopaedic and Trauma Surgery Department. Of these 45 cases, 4 were lost to follow-up over the 6 years, reducing the number of this series to 41 Ewing sarcomas in 41 different patients. The patients were reviewed retrospectively by an independent referee.
The only inclusion criterion for this study was a pathologically confirmed diagnosis of Ewing sarcoma. Tumours in the so-called Ewing sarcoma family, such as Askin tumour and peripheral primitive neuroectodermal tumour were excluded.
Once the diagnosis had been confirmed by pathological analysis of the tumour biopsy, the patients underwent a local and an extension study that comprised: computed tomography and gadolinium-based magnetic resonance of the compartment affected by the tumour, thoracic computed tomography, bone scan and bone marrow biopsy/aspirate. We use the TNM classification of the AJCC to stage the disease and subdivide the patients into two groups according to the stage at diagnosis: patients with a localised stage (AJCC stages I and II) and patients with a non-localised stage (AJCC stages III and IV).
The standard assessment of the patients included sex and date of birth, tumour site, age and stage of the sarcoma on diagnosis, type of Ewing sarcoma – bone or extraskeletal – both medical and surgical treatment received, and necrosis percentage according to Rosen and Huvos’ system dividing the patients into poor (Rosen and Huvos grades I and II) and good (grades III and IV) responders and relating them to the stage of the sarcomas.
Meetings were held of the Musculoskeletal Tumour Committee that comprised medical oncologists and radiotherapy oncologists, orthopaedic surgeons, radiologists and pathologists, all specialists in this type of tumour, who together decided on the individualised therapy for each patient according to their tumour site, stage, size and resectability, and the patient's age. Patients under the age of 18 were treated by paediatric oncology specialists following the protocol of the Spanish Society of Paediatric Oncology 2001 (SEOP 2001). Patients aged over 18 were treated according to the directives of guideline Euro-E.W.I.N.G.99. Salvage surgery was preferred to amputation, and in all cases intraoperative biopsies were performed to assess margins. Definitive therapeutic radiotherapy was chosen for unresectable tumours and for tumours whose regression after neoadjuvant treatment was not sufficient to allow surgical treatment. Postoperative radiotherapy was used for cases with involved margins after surgery and poor histological response after neoadjuvance (<10%). Preoperative radiotherapy was indicated for cases with tumour progression during neoadjuvant chemotherapy. Haematopoietic progenitor transplants were also used in paediatric patients following the directives of the SEOP 2001 guidelines. Palliative chemo and radiotherapy were used for terminal patients.
The distribution of survival was analysed according to age, sex, stage at diagnosis, type of Ewing sarcoma, axial/non axial location, involvement of margins and responses to neoadjuvant treatment. Three types of survival subgroups were distinguished based on the date of the diagnostic biopsy: during the first year (12 months or less), between the first and the fifth year (between 13 and 59 months), and from the fifth year onwards (60 months or more). The relationship between the site in the axial skeleton or extremities and the presence of metastasis at diagnosis was examined. We also studied the distribution by gender of the tumours in relation to their site, stage at diagnosis, survival and type of responder to neoadjuvant therapy. We undertook a follow-up of the natural history of the disease recording the cases that had suffered local recurrence, metastasis and metastasis together with local recurrence.
ResultsForty-one Ewing sarcomas were identified in 41 different patients, 26 males and 15 women (1.7:1) in our centre between January 2004 and December 2009. The mean age at the time of the diagnostic biopsy was 18.29 years (range 6–51) and the maximum follow-up was set at December 2015, obtaining a minimum of 72 months’ follow-up (6 years).
In terms of the stage of the disease on diagnosis, there were 28 cases in the localised subgroup (68.29%), divided into 20 males (71.42%) and 8 women (28.57%); 10 cases (24.39%) were in the non-localised group (5 males – 50% and 5 females – 50%); and we found 3 cases (7.32%), 1 male and 2 females, whose stage had not been recorded in their clinical histories that we termed “unknown stage”.
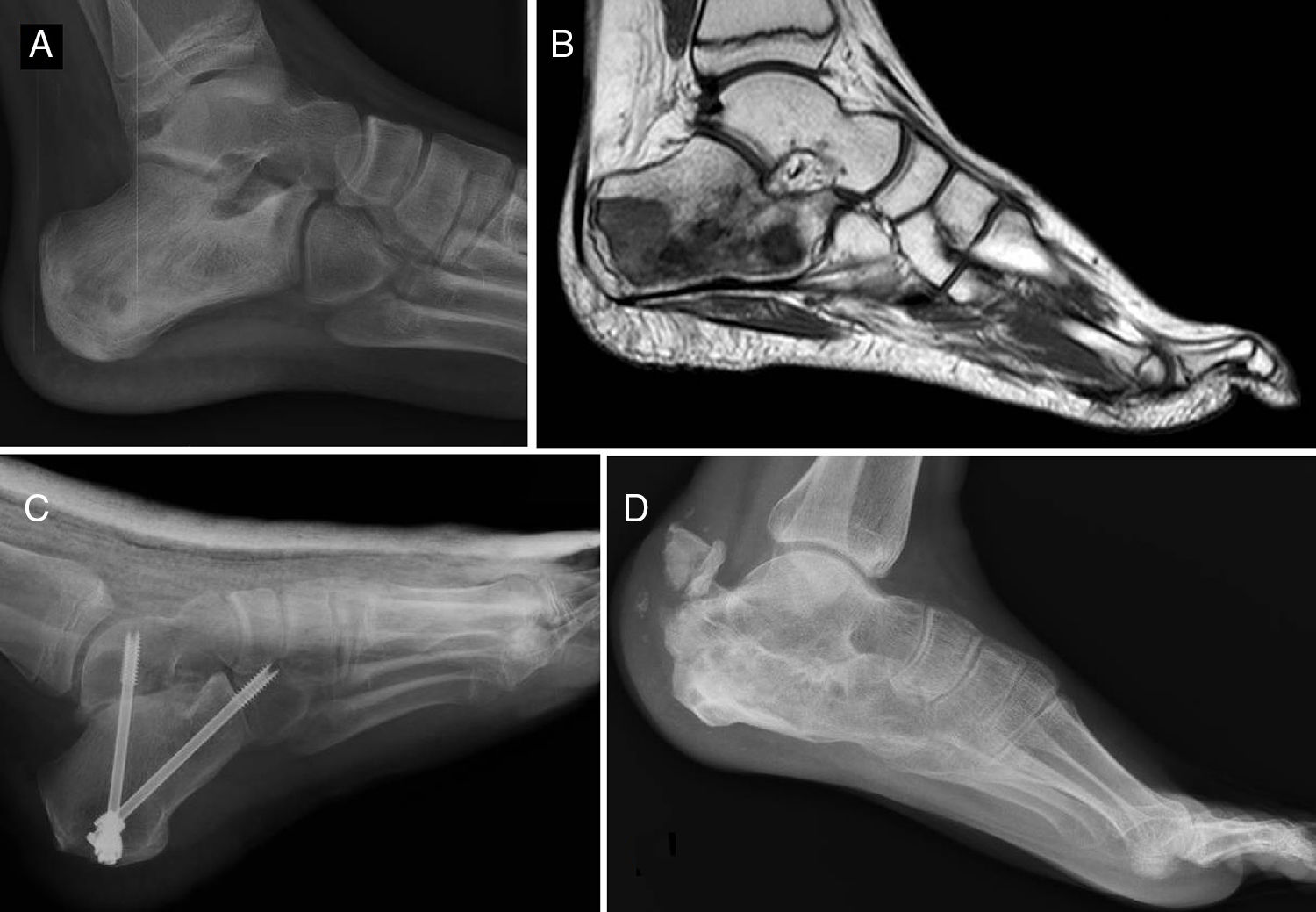
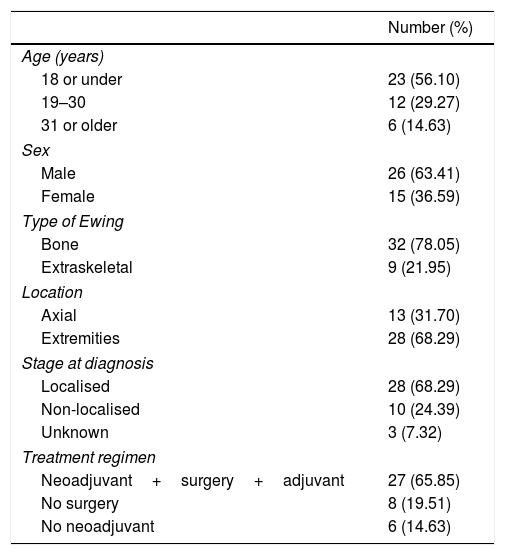
Of the total number of sarcomas, 32 (78.05%) were Ewing sarcoma of bone and the remaining 9 (21.95%) were extraskeletal Ewing tumours. The sites of the bone group were divided between: the femur (10 cases, 31.25%), spine (8 cases, 25%), fibula (4 cases, 12.5%), pelvis (3 cases, 9.38%), calcaneus (2 cases, 6.25%, Fig. 2), tibia (2 cases, 6.25%), humerus (2 cases, 6.25%), and 5th metatarsal (1 case, 3.13%). One of the patients in the bone group started with a pathological diaphyseal femur fracture, and another patient suffered a further pathological fracture, also of the femur, during the first neoadjuvant chemotherapy cycles. In the extraskeletal group, the sites were: thigh (3 cases, 33.33%), leg (2 cases, 22.22%), popliteal fossa (2 cases, 22.22%), paravertebral (1 case, 11.11%) and epidural (1 case, 11.11%). Table 1 gives a demographic summary of the patient series.
 and MR (B). Treated by resection and reconstruction with allograft (C). Radiography after 6 years’ progression (D) showing calcaneus-talus and calcaneus-cuboid consolidated arthrodesis and avulsion fraction of the posterior tuberosity which occurred in the first year, and was treated conservatively because it caused no functional impairment.")
Ewing sarcoma of the calcaneus, conventional radiology images (A) and MR (B). Treated by resection and reconstruction with allograft (C). Radiography after 6 years’ progression (D) showing calcaneus-talus and calcaneus-cuboid consolidated arthrodesis and avulsion fraction of the posterior tuberosity which occurred in the first year, and was treated conservatively because it caused no functional impairment.
Demographic summary.
| Number (%) | |
|---|---|
| Age (years) | |
| 18 or under | 23 (56.10) |
| 19–30 | 12 (29.27) |
| 31 or older | 6 (14.63) |
| Sex | |
| Male | 26 (63.41) |
| Female | 15 (36.59) |
| Type of Ewing | |
| Bone | 32 (78.05) |
| Extraskeletal | 9 (21.95) |
| Location | |
| Axial | 13 (31.70) |
| Extremities | 28 (68.29) |
| Stage at diagnosis | |
| Localised | 28 (68.29) |
| Non-localised | 10 (24.39) |
| Unknown | 3 (7.32) |
| Treatment regimen | |
| Neoadjuvant+surgery+adjuvant | 27 (65.85) |
| No surgery | 8 (19.51) |
| No neoadjuvant | 6 (14.63) |
The classical treatment regimen comprising neoadjuvant therapy followed by surgery and adjuvant therapy was followed by 27 patients (65.85%) In one of the patients, with an extraskeletal Ewing sarcoma of the thigh, a conversion to rhabdomyosarcoma was observed in the surgical specimen, which necessitated a change of treatment regimen. Eight patients (19.51%) could not undergo surgical resection because their tumours were unresectable due to their site and local spread (2 cases) or because they were multimetastatic at diagnosis (6 cases). The remaining 6 cases (14.63%) did not receive neoadjuvant treatment because 5 had axial skeletal tumours that had started with compressive symptoms requiring urgent decompression surgery, and one patient who was operated in another hospital with no prior biopsy and required surgery to expand margins prior to treatment with chemo- and radiotherapy. Eleven cases of the total 41 (26.83%) received adjuvant radiotherapy for the following reasons: 5 axial tumours that required urgent decompressive surgery prior to the diagnosis of the disease, 1 bone tumour that started with a pathological fracture, another bone tumour that suffered a pathological fracture in the first cycles of neoadjuvant chemotherapy, 2 extraskeletal tumours with poor response to neoadjuvant therapy, one with margin involvement and one metastatic progression after the surgical resection.
We found two patients with affected margins, one, with a Ewing sarcoma of bone in the proximal fibula, was treated using the classical therapeutic regimen and operated in our centre by specialist surgeons, the intraoperative biopsy was negative but the patient required surgery to expand the resection margins after the histological analysis of the surgical specimen. This patient, aged 6, had a survival of 15 months, and was classified as a poor responder using Rosen and Huvos’ system. The other case, was a patient who had undergone surgery to remove the tumour without a previous biopsy in a centre that was not a referral hospital for this disease. When Ewing sarcoma was diagnosed they were referred to our centre and underwent surgery to expand the margins and adjuvant treatment with chemotherapy. This was an extraskeletal case located in the paravertebral region in a patient aged 10 who survived for longer than the 6-year follow-up.
Of all the patients who followed the classical therapeutic regimen (27), only 26 could be classified as good or poor responders according to Rosen and Huvos’ system because this scale did not apply to the patient with the conversion to rhabdomyosarcoma. We found 9 poor responders (34.62%), divided into 7 males and 2 females (3.5:1), among whom we found 6 localised stages, one non- localised and 2 unknown stages. Seventeen were good responders (65.38%), 11 males and 6 females (1.8:1), comprising 13 localised stages, 3 non-localised and one unknown stage.
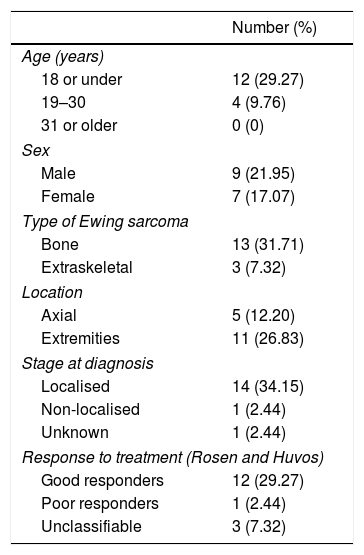
At the date set as the end of follow-up, 25 patients (60.98%), comprising 17 males and 8 females, had died as a consequence of the disease with a mean survival of 21.95 months (range .75–67 months), while 16 (39.02%), 9 males and 7 females, were still alive. Twenty-three of the patients who did not survive (92%) died during the first 5 years, 6 of them in the first year (24%), and the remaining 2 died during the sixth year. The survival rates were estimated at 85.27% at one year, and 43.90% at 5 years.
In terms of survival by age, the patients were subdivided into three age groups according to the epidemiology of this type of tumour: 18 years or under, from 19 to 30 years, and over 31 years of age. In the paediatric subgroup we found a total of 23 cases, 13.04% of the patients died during the first year, 34.7% between the first and the fifth year, and 52.17% were still alive from the fifth year. In the subgroup aged 19–30, we found a survival at 5 years of 33.33%, of 50% between the first and fifth year, and of 16.67% during the first year. In the last subgroup, we found no survivors after five years, 50% died during the first year, and the remaining 50% between the first and fifth years.
The analysis of survival according to stage at diagnosis, yielded the following data: of the 28 patients with localised sarcomas 14 died (50%), 2 during the first year, while 14 survived (50%); in the group of 10 non-localised sarcomas, we found only one survivor at 5 years (10%), 40% died during the first 12 months from the onset of the disease; in the group of unknown stage, 2 patients (66.67%) died in the first 5 years and one (33.33%) is still alive. In the subgroup of localised sarcoma patients who died, we calculated a mean survival of 25 months (range 8–67 months), and of 9.75 months in the group of non-localised sarcomas, with a range of .75–29 months.
With regard to survival according to axial/non axial location, we found 13 axial cases and 28 non axial. Among the axial cases, we calculated a survival rate of 38.46% from the fifth year of follow-up, of 30.77% between the first and fifth years, and of 30.77% in the first twelve months of follow-up. In the non axial subgroup, mortality during the first year rose to 14.29%, 46.43% survived between the first and the fifth year, and 39.29% were still alive at the fifth year. We performed a subanalysis of the axial cases differentiating the pelvic cases (8 cases, 61.54%) from the extrapelvic cases (5 cases, 38.46%), and we found that all the patients with pelvic disease died before the end of follow-up, half of them during the first year and the other half before the 2nd year. All the axial extrapelvic cases, where we found an epidural tumour, a paravertebral tumour and three bone tumours in vertebral bodies D11, L2 and L3, had survived at the end of follow-up.
With regard to survival according to the type of Ewing sarcoma, bone or extraskeletal, we found that 13 of the 32 Ewing sarcomas of bone (40.63%) had survived the entire 6 years of follow-up, 17 (53.13%) died in the first 5 years of the disease (4 in the first year—12.5%–), and the 2 remaining cases (6.25%) died during the sixth year of follow-up. In the extraskeletal group, 6 of the 9 (66.67%) died in the first 5 years of follow-up, 2 of the 6 during the twelve first months, and only 3 patients (33.33%) had survived to the end of follow-up.
In terms of response to neoadjuvant therapy, 11 of the good responders (64.71%) had survived throughout the follow-up, and the remaining 6 (35.29%) died no earlier than 14 months since onset of the disease. None of the patients in the poor responder group survived. Table 2 gives a summary of the most relevant survival data.
Survival at 5 years.
| Number (%) | |
|---|---|
| Age (years) | |
| 18 or under | 12 (29.27) |
| 19–30 | 4 (9.76) |
| 31 or older | 0 (0) |
| Sex | |
| Male | 9 (21.95) |
| Female | 7 (17.07) |
| Type of Ewing sarcoma | |
| Bone | 13 (31.71) |
| Extraskeletal | 3 (7.32) |
| Location | |
| Axial | 5 (12.20) |
| Extremities | 11 (26.83) |
| Stage at diagnosis | |
| Localised | 14 (34.15) |
| Non-localised | 1 (2.44) |
| Unknown | 1 (2.44) |
| Response to treatment (Rosen and Huvos) | |
| Good responders | 12 (29.27) |
| Poor responders | 1 (2.44) |
| Unclassifiable | 3 (7.32) |
Of all the sarcomas analysed, 9 were metastatic on diagnosis (21.95%), 6 were bone –4 in the axial skeleton and 2 in the extremities– and 3 extraeskeletal –all in the extremities. Putting these data to a further analysis we found that of a total of 13 Ewing sarcomas sited on the extremities, 5 (17.86%) were stage IV on diagnosis.
Of the total 41 patients, 13 progressed towards a cure of the disease, with no local recurrences or metastasis during the 6 months of follow-up (31.71%). Two patients, 4.88%, suffered local recurrence that was treated according to the abovementioned protocol. Twenty-one point ninety-five percent (9 patients) developed metastases with no recurrences, essentially bone and lung, pleural in one case and lymph node in the other. Seven patients, 17.07%, suffered local recurrences together with lymph node, bone and lung metastases, we found one case with liver metastases. Eight patients (19.51%) started with metastatic lung and/or bone disease, all of them progressed to early development of other metastases (brain in one case, pleural in another). One patient developed a B-cell lymphoma secondary to adjuvant radiotherapy. In one case the tumour progressed to rhabdomyosarcoma on the pathological analysis of the surgical specimen, subsequently developing systemic metastases.
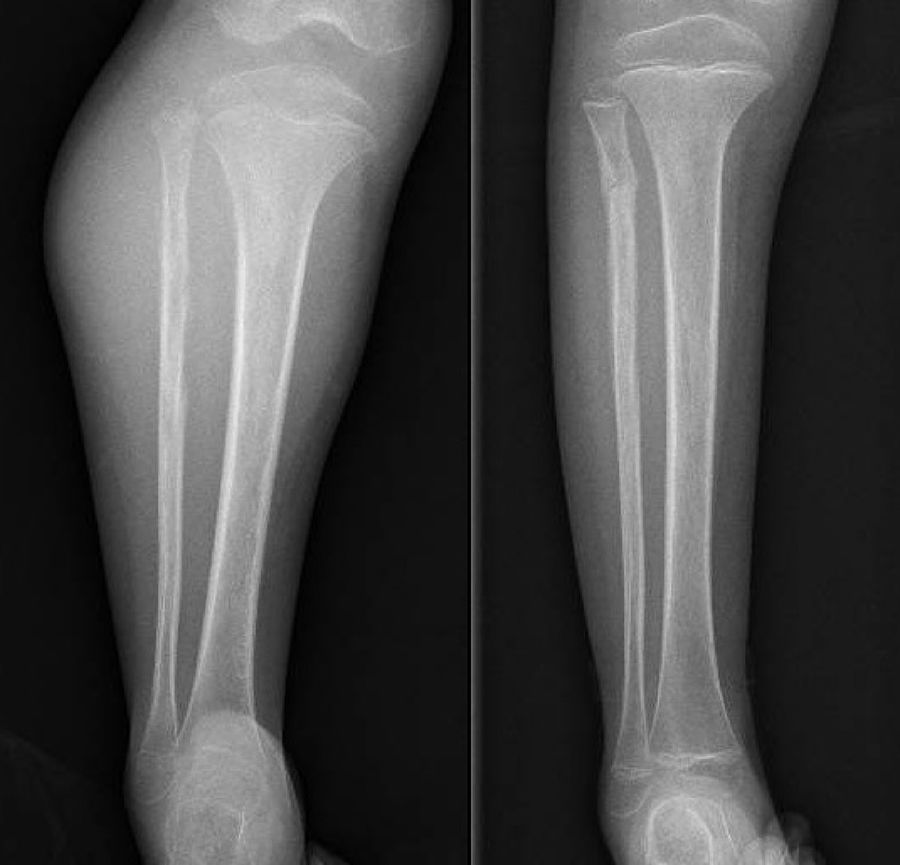
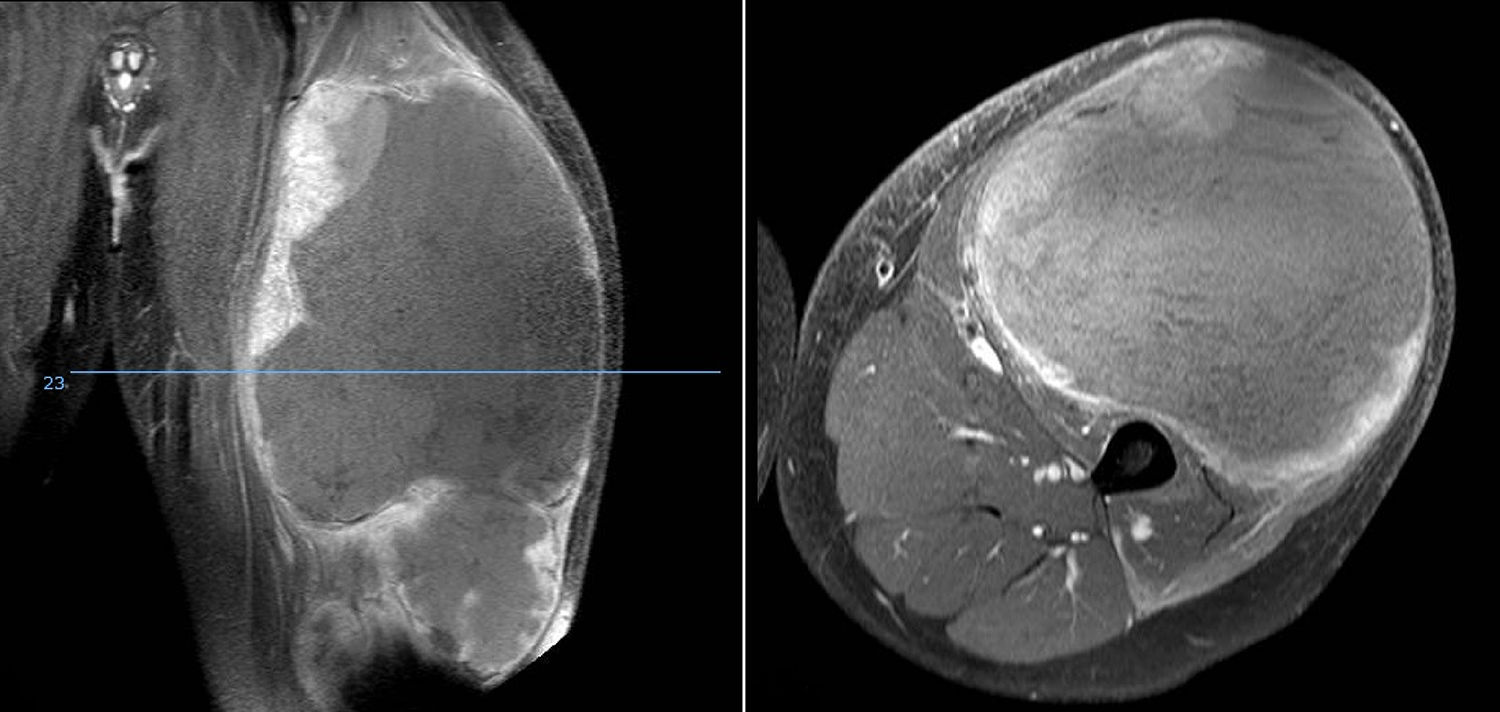
DiscussionWe analysed numerous factors relating to Ewing sarcoma in a series of 41 patients treated in the same hospital in Spain over 6 years. In our series, 2 thirds of the patients were males and a third females, almost 70% were under the age of 20 at the time of pathological diagnosis of the tumour, this data matches that in the literature.1,4,7,8 It is rarely diagnosed in children under 5 or people over the age of 30,3,13 we found five cases in our series (12.2%) in this age range: one patient aged 51 and another of 49 with extraskeletal forms (thigh –Figure 3– and leg respectively), and a further three patients of 31, 32 and 35 years of age with Ewing sarcoma of the bone in the tibia and femur respectively (Fig. 4).


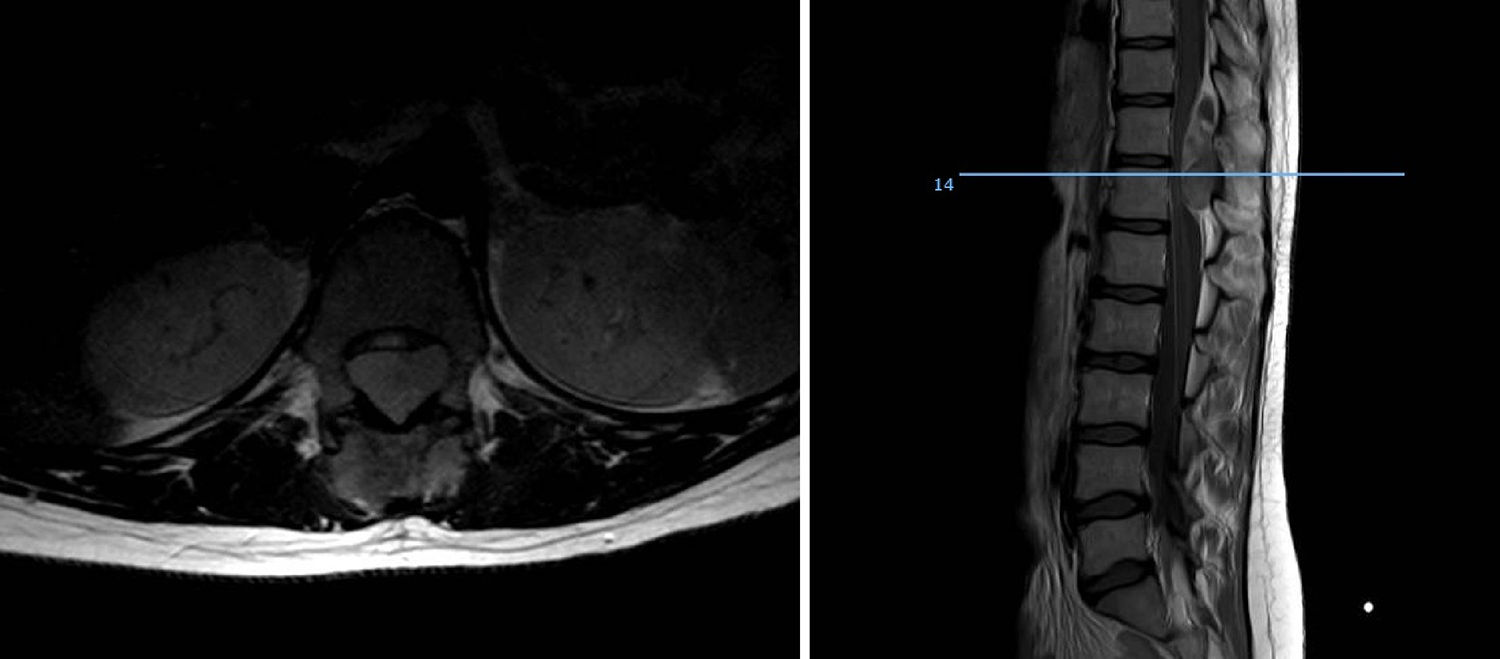
In terms of site, the diaphysis or metaphysodiaphyseal segments of the long bones, especially the femur, are the most common sites for this sarcoma. In our series almost half the total tumours arose in these sites (18 cases), the femur being the most common bone, followed by the fibula, tibia and humerus. This disease affects the flat bones more frequently than other bone sarcomas, a fourth of our patients presented with sarcoma in the axial skeleton, 3 in the pelvis and 8 in the vertebral spine, the sacrum being the most common sublocation of this group. Up to 20% were extraskeletal sarcomas, soft tissue tumours with no bone involvement but with the same histology as Ewing sarcoma of bone, mainly arising in the paravertebral region, thoracic wall and lower limbs. Our analysis found 21.95% extraskeletal cases, the majority of which were distributed in the lower limbs, and in paravertebral and epidural regions (2 cases). We found no case of epiphyseal location, and only one of the patients started with a pathological fracture, which is in line with the current literature.
Twenty-one percent of our patients had metastasis at time of diagnosis of the disease, which is in line with the 15–20% described in different publications. Almost half the cases that were metastatic on diagnosis, had an axial Ewing sarcoma, either bone or extraskeletal; 30% of those with an axial Ewing sarcoma had metastases on diagnosis, compared to 17% of those with a Ewing sarcoma in the extremities. In other words, in our series the patients with axial disease had almost double the likelihood of being stage IV at diagnosis than those with a Ewing sarcoma located in the extremities.
Survival rates have increased from 10–15% to 65–70% over the past 40 years. This improvement can essentially be put down to the aggressive use of chemotherapy and multidisciplinary treatment.4,14,15 However, the prognosis is still poor, especially for patients with disseminated disease or early recurrence, whose disease-free survival at 5 years is 9–30%. In our series, we calculated an overall survival rate of 85.27% in the first year and of 43.90% at 5 years, and a mean survival of those who died of 21.95 months (range 0.75–67 months).
It seems that age is a prognostic factor in this disease, however there is still debate in the literature about this.6,10,16,17 In our study, the analysis of survival according to age highlights that the best figures are obtained in paediatric patients, 52.17% of the total paediatric subgroup were still alive from the fifth year of follow-up, 85% of these survivors were 14 years of age or under. With regard to the complete series (43.90% survival at 5 years), this implies that almost 30% of this 43.90% of survivors were patients aged 18 or under at the time of diagnosis. Furthermore, it was in patients aged over 30, where this type of tumour is uncommon, where we collected the worst survival rates: we found no survivals at 5 years of follow-up, half died during the first year, and the other half between the first and fifth years. In the intermediate age group, we found 4 patients who survived longer than 5 years (33.33%), 2 with a survival under one year, and 6 (50%) who died between the first and fifth years of follow-up. It has been postulated that the biology of the disease differs according to the different age groups, that the disease is more aggressive in older patients or that their response to the current treatments is poorer.10
Classifying our patients according to their presentation stage, half the localised-stage sarcomas survived throughout the follow-up, whereas the other half died as a consequence of the disease, 14.28% during the first year, with a mean survival rate of those who died of 25 months (range 8–67 months). However, in the localised-stage sarcoma group we found only one survivor at 5 years (10%), 40% died during the first year of the disease, with a mean survival rate of those who died of 9.75 months. Our survival data are lower than those reported in the current literature.
The tumour arising in the pelvis is a poor prognostic factor for many authors, such as Bacci et al.18 who describe in their 2003 article on non-metastatic pelvic Ewing sarcomas that the prognosis for patients with pelvic disease is poorer than that of the extrapelvic cases. They suggest that this might be because a tumour located in the pelvis makes local treatment difficult which results in high recurrence rates. In our study we had 13 axial tumours, 8 of which were pelvic. All the pelvic Ewing sarcoma cases died before the end of follow-up, half during the first year and the other half between the first and the fifth year, not one patient survived longer than 24 months. The axial extrapelvic cases had a 100% survival rate from the fifth year of follow-up.
Applenbaum et al.19 analysed extraskeletal Ewing sarcomas in their article published in the journal Cancer in 2011 and reached the conclusion that these tumours had a poorer prognosis in the first years of the disease because the disease had been diagnosed later and had appeared at an older age compared with the Ewing sarcomas of bone. Although our number of cases did not enable us to obtain statistically significant results, we must mention that in our series we obtained better survival of Ewing sarcomas of bone than of the extraskeletal sarcomas (40.63% and 33.33% overall survival, respectively), and that the two older patients in the series had an extraskeletal form of the disease, which coincides with the data of the abovementioned publication.
Most authors agree that patients with a good histological response to neoadjuvant therapy have a better prognosis than the poor responders.1,3,6 Our series supports this claim: of the total survivors at the end of follow-up – 16 patients – 11 belonged to the good responder subgroup and the remaining 5 did not receive neoadjuvant therapy for the abovementioned reasons, therefore they could not be classified according to Rosen and Huvos’ system. We found no survivals at the end of follow-up among the poor responders.
In terms of margins, we believe that we can reach no conclusions in our study because only one correctly-treated patient had to undergo surgery to expand the margins and died at 15 months; this patient was in the poor responder group. The other patient was operated in a centre that was not a referral hospital and with no previous biopsy, and was referred to our centre for multidisciplinary, individualised treatment because the anatomic pathology results of the resected tumour showed the presence of small and round cells.
To conclude, Ewing sarcoma is more common in males and still carries high mortality despite medical and surgical advances. This is why some authors consider it a disseminated disease from the outset with micro-metastases that cannot be detected with the current diagnostic techniques, and it is these that really determine survival of the disease. The femoral metaphysodiaphyseal region is the most affected, and the sacrum is the most common site for axial tumours. The extraeskeletal forms and the patients who were poor responders according to Rosen and Huvos’ system have not only higher but also earlier rates of mortality compared to the other forms. Pelvic Ewing sarcomas in general are diagnosed late which makes them more likely to be metastatic at the time of diagnosis, which worsens their prognosis. The best tools currently at our disposal to offer patients the greatest possibilities of a cure are early diagnosis and multidisciplinary therapy in referral centres.
Level of evidenceLevel of evidence IV.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Borrego-Paredes E, Prada-Chamorro E, Chacón-Cartaya S, Santos-Rodas A, Gallo-Ayala JM, Hernández-Beneit JM. Sarcoma de Ewing, análisis de supervivencia a los 6 años con terapia multidisciplinar. Rev Esp Cir Ortop Traumatol. 2019;63:86–94.
