




El objetivo de este estudio es presentar nuestra serie de casos de sarcoma de Ewing y los datos de supervivencia obtenidos a medio plazo utilizando un protocolo de terapia multidisciplinar.
Material, método y resultadosCuarenta y un sarcomas de Ewing fueron diagnosticados, tratados y evolucionados en nuestro centro entre 2004 y 2009 con una media de edad de 18,29 años. Hasta un 78% correspondieron a Ewing óseo siendo el fémur la localización más frecuente. El 68% presentó un estadio localizado en el momento del diagnóstico. Al final del seguimiento la supervivencia no llegó al 40% de los pacientes, falleciendo la mayoría en los primeros 5 años de seguimiento.
DiscusiónEn España, el sarcoma de Ewing es el tumor óseo maligno primario más frecuente en la infancia, por delante del osteosarcoma. Su tasa de supervivencia ha aumentado mucho en los últimos 40 años, mejoría atribuible fundamentalmente al uso agresivo de la quimioterapia y al tratamiento mutidisciplinar, pero su pronóstico sigue siendo muy pobre sobre todo en aquellos que presentan metástasis al diagnóstico, principal factor pronóstico adverso. Dada su alta mortalidad, son muchos los autores que lo consideran como una enfermedad diseminada desde el principio, con micrometástasis no detectables que son las que condicionan la supervivencia final.
ConclusionesEl diagnóstico precoz y el tratamiento multidisciplinar en centros de referencia son las mejores estrategias con las que contamos en la actualidad para proporcionarles a los pacientes las máximas posibilidades de curación de esta enfermedad.
The purpose of this study is to present our series of Ewing sarcoma cases and the survival data obtained in the medium term, using a multidisciplinary therapy protocol.
Material, methods and resultsForty-one Ewing sarcomas were diagnosed, treated and followed-up in our hospital between 2004 and 2009 with an average age of 18.29 years. Seventy-eight percent were to Ewing sarcoma of the bone, the femur being the most frequent location. Sixty-eight percent had a localized stage at the time of diagnosis. At the end of follow-up, 40% of the patients did not survive, most died within the first 5 years of follow-up.
DiscussionIn Spain, Ewing sarcoma is the most common primary malignant bone tumour in childhood, ahead of osteosarcoma. Its survival rate has increased greatly in the last 40 years, improvement attributable mainly to the aggressive use of chemotherapy and to multidisciplinary treatment, but its prognosis remains very poor, especially for those with metastasis at diagnosis, the main adverse prognostic factor. Because of its high mortality, many authors consider it a disseminated disease from the beginning, with non- detectable micrometastasis that condition final survival.
ConclusionsEarly diagnosis and multidisciplinary therapy in referral centres are the best strategies currently available to us to provide these patients the maximum possibilities of cure of this disease.
El sarcoma de Ewing es un tumor primario maligno primitivo del hueso descrito por James Ewing en 1921, quien lo llamó endotelioma difuso del hueso1–3. Su histogénesis exacta es desconocida, parece que deriva de células de la médula ósea4 correspondiendo a una forma pobremente diferenciada del tumor neuroectodérmico primitivo2. Se trata de una neoplasia de células azules redondas pequeñas con una translocación cromosómica común que implica, en casi todos los casos, el gen EWSR1 en el cromosoma 22 y a un miembro de la familia de factores de transcripción ETS en el cromosoma 91,3,5.
El Ewing es el segundo tumor óseo maligno primario más frecuente en la infancia, por detrás del osteosarcoma1,6, y el más frecuente en España, según el registro nacional de cáncer pediátrico de 20166. El 80% aparece en menores de 20 años, es infrecuente en menores de 5 años y en mayores de 301, es más frecuente en varones y excepcional en la raza negra. Su incidencia anual aproximada es de 3 casos por millón de individuos de raza blanca y menores de 21 años1,4,7,8.
Su localización más frecuente es en diáfisis o segmentos metafisodiafisarios de huesos largos, siendo el fémur el asiento del 20-27%3,8, seguido de tibia, peroné y húmero. A diferencia del osteosarcoma, aparece con frecuencia en los huesos planos del esqueleto axial, fundamentalmente en pelvis, pared torácica (en este caso se denomina tumor de Askin) y en sacro1,3. Hasta un 20% son sarcomas extraesqueléticos, tumores de partes blandas sin afectación ósea pero con la misma histología que el Ewing óseo, asentando sobre todo a nivel paravertebral, pared torácica y en miembros inferiores1,9. La localización a nivel de sistema nervioso central es rara, tanto primaria como metastásica, generalmente se trata de Ewing extraesqueléticos epidurales.
Tumoración palpable dolorosa (fig. 1) junto con enrojecimiento y calor constituye la forma de presentación más frecuente. Hasta una quinta parte presenta además síntomas constitucionales debidos a la liberación de citoquinas10 tales como fiebre, astenia, anorexia y pérdida de peso1,3, que incluso pueden inducir al diagnóstico erróneo de una osteomielitis4. El estadio más frecuente de presentación es el IIB de la American Joint Committe on Cancer (AJCC)4 y alrededor de un 20% presentan metástasis al diagnóstico, siendo el pulmón la localización más frecuente, pero se estima que muchos más presentan micrometástasis al diagnóstico que no pueden ser detectadas con las técnicas diagnósticas actuales. Estas serían las responsables del mal pronóstico de los Ewing catalogados como «localizados»3,10,11.

Radiológicamente aparece como una lesión osteolítica con patrón apolillado o permeativo, reacción perióstica en piel de cebolla y una gran masa de partes blandas no mineralizada. En huesos planos, predomina el patrón mixto lítico-blástico con un área de esclerosis1,4,9,12.
El tratamiento del sarcoma de Ewing es multidisciplinar siendo la quimioterapia la base y la resección quirúrgica el pilar fundamental para el control local del mismo. Aunque clásicamente cirugía y radioterapia han estado al mismo nivel en cuanto a control local, actualmente se sabe que la cirugía aporta mejores resultados en supervivencia libre de enfermedad que la radioterapia sola3,4,10 evitando además el riesgo de sarcomas secundarios radioinducidos4. Así, en pacientes sin evidencia de enfermedad metastásica en el momento del diagnóstico, los protocolos para el tratamiento del sarcoma de Ewing incorporan poliquimioterapia pre- y postoperatoria –se sabe desde los años 70 que la quimioterapia mejora la supervivencia y que la asociación de varios fármacos es superior a la monoterapia1–, resección quirúrgica y radioterapia en el caso de márgenes afectos o de recidiva1,4. Se reserva la radioterapia aislada para lesiones grandes e irresecables axiales con el objeto de evitar cirugías mutilantes3 y para pacientes con metástasis pulmonares que hayan tenido una respuesta favorable a la quimioterapia, incluso cuando se haya obtenido una remisión completa, ya que la tasa de recaída pulmonar puede reducirse hasta en un 50% con el uso de radioterapia1. La quimioterapia intensiva y la megaterapia con trasplante autólogo de células madre hematopoyéticas son buenas alternativas para el tratamiento de pacientes de riesgo1,6. Las innovaciones en el tratamiento de este tumor inluyen nuevos quimioterápicos, destacando gemcitabina-docetaxel, trabectedina, anticuerpos anti-IGF1R, fenretinide, sirolimus y deforolimus3.
Las tasas de supervivencia han aumentado de un 10-15% hasta un 65-70% en los últimos 40 años3, mejoría fundamentalmente debida al uso agresivo de la quimioterapia y al tratamiento multidisciplinar4,6. Sin embargo, el pronóstico sigue siendo pobre sobre todo en pacientes con enfermedad diseminada o recidiva precoz1,3,4,6 siendo la supervivencia libre de enfermedad a los 5 años de un 9-30%10. Los factores pronósticos más importantes son la presencia de metástasis al diagnóstico, lo cual constituye el principal factor pronóstico adverso, la localización del tumor y el tamaño de la lesión, la carga tumoral y la respuesta biológica a la quimioterapia neoadyuvante y el tiempo transcurrido desde el diagnóstico hasta la recidiva1,3. Otros factores en el momento del diagnóstico como edad mayor de 14 años, niveles elevados de LDH y corta duración de los síntomas previos parecen también relacionados con un pronóstico pobre de la enfermedad6.
Material y métodoRealizamos un análisis retrospectivo, aprobado por la Junta de Revisión Institucional, entre los años 2004 y 2009 (72 meses) de los sarcomas de Ewing diagnosticados en nuestro hospital, centro de referencia de tumores musculoesqueléticos de las comunidades autónomas de Andalucía, Islas Canarias y Extremadura. Con el objetivo de obtener un periodo de seguimiento de 6 años, establecimos el final del seguimiento en diciembre de 2015. Durante este tiempo, 45 sarcomas de Ewing en 45 pacientes distintos fueron diagnosticados mediante una búsqueda conjunta realizada entre el Departamento de Anatomía Patológica y la Unidad de Tumores Musculoesqueléticos del servicio de Cirugía Ortopédica y Traumatología. De esos 45 casos, 4 fueron pérdida de seguimiento a lo largo de esos 6 años, reduciendo el número de esta serie a 41 sarcomas de Ewing en 41 pacientes distintos. Los pacientes fueron revisados de forma retrospectiva por un revisor independiente.
El único criterio de inclusión para este estudio fue la confirmación anatomopatológica del diagnóstico de sarcoma de Ewing. Se excluyeron aquellos tumores incluidos en la denominada familia del sarcoma de Ewing tales como el tumor de Askin y el tumor neuroectodérmico primitivo periférico.
Una vez obtenida la confirmación diagnóstica mediante análisis anatomopatológico de la biopsia de la lesión, los pacientes fueron sometidos a un estudio local y de extensión constituido por: tomografía computarizada y resonancia magnética con gadolinio del compartimento afecto por el tumor, tomografía computarizada torácica, gammagrafía ósea y biopsia/ aspirado de médula ósea. Utilizamos la clasificación TNM de la AJCC para la estadificación de la enfermedad y subdividimos a los pacientes en dos grupos en función del estadio al diagnóstico: pacientes en estadio localizados (I y II de la AJCC) y pacientes en estadio no localizado (III y IV de la AJCC).
La evaluación estándar de los pacientes incluyó sexo y fecha de nacimiento de los mismos, localización del tumor, edad del paciente y estadio del sarcoma al diagnóstico, tipo de Ewing –óseo o extraesquelético–, tratamiento recibido tanto médico como quirúrgico y porcentaje de necrosis según la escala de Rosen y Huvos dividiendo a los pacientes en malos (grados I y II de Rosen y Huvos) y buenos respondedores (grados III y IV) y relacionándolos con el estadio de los sarcomas.
Con respecto al tratamiento de los pacientes, fue en reuniones del Comité de Tumores Musculoesqueléticos, compuestas por oncólogos médicos y oncólogos radioterápicos, cirujanos ortopédicos, radiológos y anatomopatólogos, todos ellos especialistas en este tipo de tumores, donde se decidió conjuntamente la terapia individualizada para cada paciente según la localización del tumor, su estadio, su tamaño y resecabilidad y la edad del paciente. Los menores de 18 años fueron tratados por especialistas en Oncología Pediátrica siguiendo el protocolo de la Sociedad Española de Oncología Pediátrica de 2001 (SEOP 2001). Los mayores de 18 años, fueron tratados siguiendo las directrices de la guía Euro-E.W.I.N.G.99. Se prefirió la cirugía de salvamento en lugar de las amputaciones, y en todos los casos se realizaron biopsias intraoperatorias para la valoración de los márgenes. Se optó por radioterapia terapéutica definitiva en tumores irresecables y en tumores cuya regresión tras el tratamiento neoadyuvante no fuera suficiente como para permitir la resección quirúrgica de los mismos. La radioterapia postoperatoria se utilizó en casos de márgenes afectos tras cirugía y respuesta histológica pobre tras neoadyuvancia (<10%). La radioterapia preoperatoria se indicó en aquellos casos con progresión tumoral durante la quimioterapia neoadyuvante. En pacientes pediátricos se realizaron además trasplantes de progenitores hematopoyéticos siguiendo las directrices de la guía SEOP 2001. Se empleó quimio- y radioterapia paliativas en casos terminales.
Se analizó la distribución de la supervivencia según edad, sexo, estadio al diagnóstico, tipo de Ewing, localización axial/ no axial, afectación de márgenes y respuestas al tratamiento neoadyuvante. Se distinguieron tres subgrupos de supervivencia en los mencionados análisis, partiendo de la fecha en la que la biopsia diagnóstica fue realizada: durante el primer año (12 meses o menos), entre el primer y el quinto año (entre 13 y 59 meses) y a partir del quinto año (60 meses o más). Se examinó la relación entre la localización en esqueleto axial o extremidades y la presencia de metástasis al diagnóstico. Se estudió además la distribución por géneros de los tumores en relación con su localización, estadio al diagnóstico, supervivencia y tipo de respondedor a la terapia neoadyuvante. Realizamos un seguimiento de la historia natural de la enfermedad registrando aquellos casos que sufrieron recidivas locales, metástasis y metástasis junto con recidivas locales.
ResultadosCuarenta y un sarcomas de Ewing en 41 pacientes distintos, 26 varones y 15 mujeres (1,7:1), fueron identificados en nuestro centro entre enero de 2004 y diciembre de 2009. La edad media en el momento de la biopsia diagnóstica fue de 18,29 años (rango 6-51) y el seguimiento máximo se estableció en diciembre de 2015, obteniendo un mínimo de 72 meses de seguimiento (6 años).
En lo que respecta al estadio de la enfermedad al diagnóstico, 28 casos constituían el subgrupo localizado (68,29%), divididos en 20 varones (71,42%) y 8 mujeres (28,57%); 10 casos (24,39%) el no localizado (5 varones –50%– y 5 mujeres –50%–); y encontramos 3 casos (7,32%), 1 varón y 2 mujeres, con estadio no recogido en la historia clínica los cuales denominamos «estadio no conocido».
Del total de sarcomas, 32 (78,05%) correspondieron a Ewing de tipo óseo y los 9 restantes (21,95%) a Ewing extraesquelético. Las localizaciones del grupo óseo se repartieron entre: fémur (10 casos, 31,25%), columna (8 casos, 25%), peroné (4 casos, 12,5%), pelvis (3 casos, 9,38%), calcáneo (2 casos, 6,25% figura 2), tibia (2 casos, 6,25%), húmero (2 casos, 6,25%) y 5.° metatarsiano (1 caso, 3,13%). Uno de los pacientes del grupo óseo debutó con una fractura patológica a nivel diafisario femoral, y otro paciente sufrió otra fractura patológica también femoral durante los primeros ciclos de quimioterapia neoadyuvante. En lo que al grupo extraesquelético se refiere, los emplazamientos fueron: muslo (3 casos, 33,33%), pierna (2 casos, 22,22%), hueco poplíteo (2 casos, 22,22%), paravertebral (1 caso, 11,11%) y epidural (1 caso, 11,11%). En la tabla 1 se expone un resumen demográfico de la serie de pacientes.
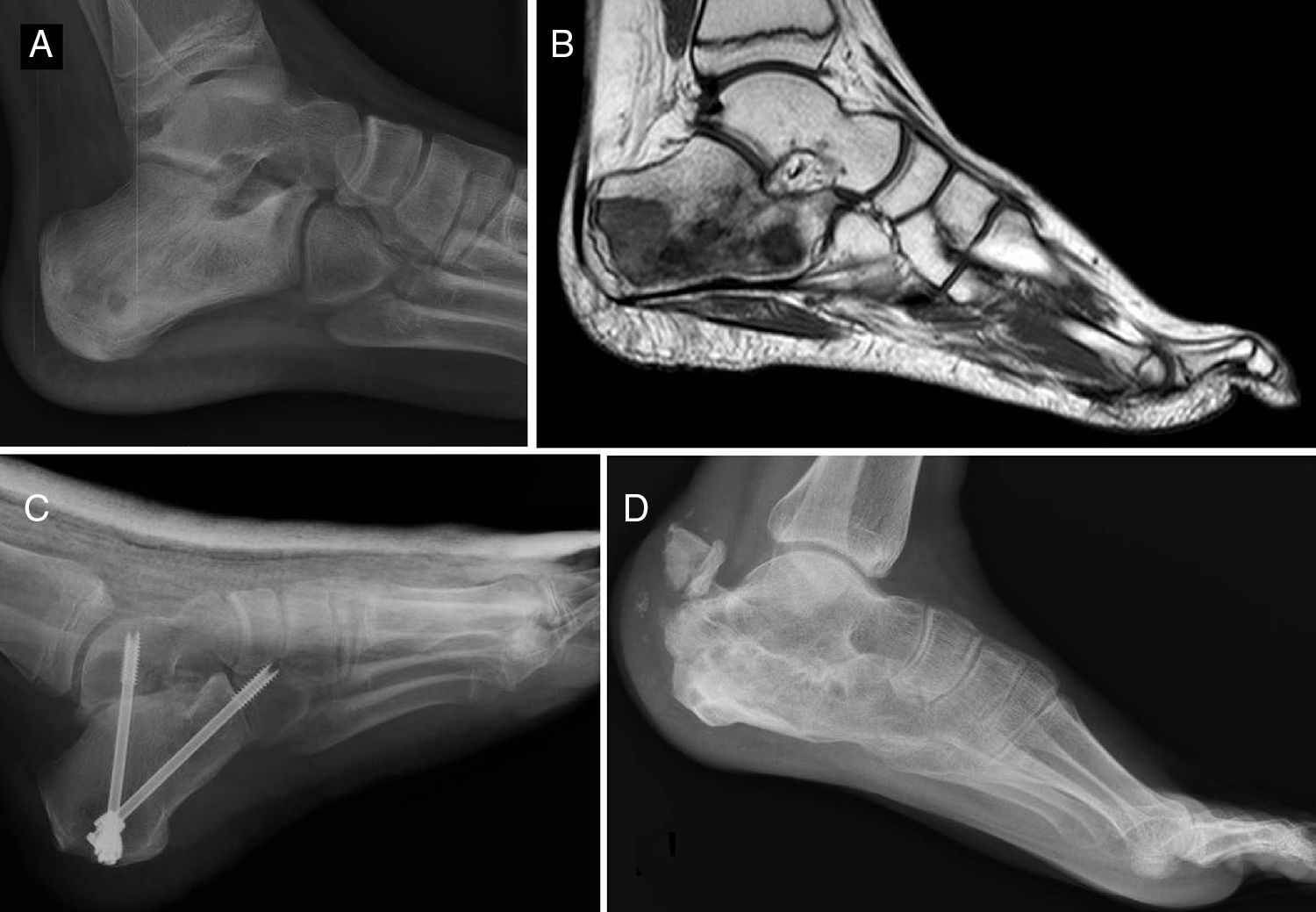
 y de RM (B). Fue tratado mediante resección y reconstrucción con aloinjerto (C). Radiografía tras 6 años de evolución (D) en la que se aprecia artrodesis consolidada calcáneo-astragalina y calcáneo-cuboidea y fractura-avulsión de tuberosidad posterior ocurrida en el primer año tratada de manera conservadora porque no generó alteración funcional.")
Sarcoma de Ewing de calcáneo, imágenes de radiología convencional (A) y de RM (B). Fue tratado mediante resección y reconstrucción con aloinjerto (C). Radiografía tras 6 años de evolución (D) en la que se aprecia artrodesis consolidada calcáneo-astragalina y calcáneo-cuboidea y fractura-avulsión de tuberosidad posterior ocurrida en el primer año tratada de manera conservadora porque no generó alteración funcional.
Resumen demográfico
| Número (%) | |
|---|---|
| Edad (años) | |
| 18 o menos | 23 (56,10) |
| 19-30 | 12 (29,27) |
| 31 o más | 6 (14,63) |
| Sexo | |
| Masculino | 26 (63,41) |
| Femenino | 15 (36,59) |
| Tipo de Ewing | |
| Óseo | 32 (78,05) |
| Extraesqueléticos | 9 (21,95) |
| Localización | |
| Axial | 13 (31,70) |
| Extremidades | 28 (68,29) |
| Estadio al diagnóstico | |
| Localizado | 28 (68,29) |
| No localizado | 10 (24,39) |
| Desconocido | 3 (7,32) |
| Esquema de tratamiento | |
| Neoadyuvante + cirugía + adyuvante | 27 (65,85) |
| Sin cirugía | 8 (19,51) |
| Sin neoadyuvancia | 6 (14,63) |
El esquema clásico de tratamiento consistente en terapia neoadyuvante seguida de cirugía y de terapia adyuvante fue seguido por 27 pacientes (65,85%), en uno de ellos, un Ewing extraesquelético de muslo, se objetivó conversión a rabdomiosarcoma en la pieza de resección lo cual obligó a modificar el esquema de tratamiento. Ocho pacientes (19,51%) no pudieron someterse a resección quirúrgica al tratarse de tumores irresecables por su localización y extensión local (2 casos) o de pacientes multimetastásicos al diagnóstico (6 casos). Los 6 casos restantes (14,63%) no recibieron tratamiento neoadyuvante debido a que 5 de ellos fueron tumores del esqueleto axial que debutaron con síntomas compresivos requiriendo de cirugía urgente descompresiva, y un paciente que fue intervenido en otro hospital sin biopsia previa y precisó de una cirugía de ampliación de márgenes previa al tratamiento con quimio- y radioterapia. En 11 casos del total de 41 (26,83%) se empleó radioterapia adyuvante por las siguientes razones: 5 tumores axiales que precisaron de cirugía urgente descompresiva previa al diagnóstico de la enfermedad, 1 tumor óseo que debutó con una fractura patológica, otro tumor óseo que sufrió una fractura patológica en los primeros ciclos de quimioterapia neoadyuvante, 2 tumores extraesqueléticos con escasa respuesta a la terapia neoadyuvante, una afectación de márgenes y una progresión metastásica tras la resección quirúrgica.
Encontramos a dos pacientes con márgenes afectos, uno de ellos, afecto de un Ewing óseo localizado en peroné proximal, fue tratado según el esquema terapéutico clásico e intervenido en nuestro centro por cirujanos especializados, la biopsia intraoperatoria fue negativa pero precisó de una cirugía de ampliación de márgenes tras el análisis anatomopatológico de la pieza de resección. Este paciente, de 6 años de edad, obtuvo una supervivencia de 15 meses, y fue clasificado como mal respondedor según la escala de Rosen y Huvos. El otro caso, resultó ser un paciente que fue sometido a una cirugía de exéresis de la tumoración sin biopsia previa en un centro no de referencia para esta enfermedad. Ante el diagnóstico de sarcoma de Ewing, fue derivado a nuestro centro donde se sometió a cirugía de ampliación de márgenes y a tratamiento adyuvante con quimioterapia. Se trató de un caso extraesquelético de localización paravertebral en un paciente de 10 años cuya supervivencia superó los 6 años de seguimiento.
De todos los pacientes que siguieron el esquema terapéutico clásico (27), solo 26 pudieron ser clasificados en buenos y malos respondedores siguiendo la escala de Rosen y Huvos debido a que en el paciente de la conversión a rabdomiosarcoma no fue aplicable dicha escala. Encontramos 9 malos respondedores (34,62%), divididos en 7 varones y 2 mujeres (3,5:1), entre los que se encontraban 6 estadios localizados, uno no localizado y 2 estadios desconocidos. Diecisiete fueron los buenos respondedores (65,38%), 11 varones y 6 mujeres (1,8:1), conformados por 13 estadios localizados, 3 no localizados y un estadio desconocido.
En la fecha establecida como final del seguimiento, 25 pacientes (60,98%) constituidos por 17 varones y 8 mujeres habían fallecido a consecuencia de la enfermedad con una media de supervivencia de 21,95 meses (0,75-67 meses de rango), mientras que 16 (39,02%), 9 varones y 7 mujeres, permanecían vivos. Veintitrés de los pacientes fallecidos (92%) lo hicieron durante los 5 primeros años, 6 de ellos en el primer año (24%), y los 2 restantes fallecieron durante el sexto año. Las tasas de supervivencia se estimaron en 85,27% al año y en 43,90% a los 5 años.
En cuanto a la supervivencia por edades, los pacientes fueron subdivididos en tres grupos de edad en función de la epidemiología de este tipo de tumor: 18 años o menores, de 19 a 30 años y mayores de 31 años. En el subgrupo pediátrico encontramos un total de 23 casos, un 13,04% de los pacientes fallecieron durante el primer año, un 34,7% entre el primer y el quinto año y un 52,17% continuaron con vida a partir del quinto año. En el subgrupo de 19-30 años de edad, encontramos una supervivencia a los cinco años del 33,33%, al 50% entre el primer y el quinto año y del 16,67% durante el primer año. En el último subgrupo, no hallamos supervivientes a partir de los cinco años, un 50% falleció durante el primer año y el 50% restante entre el primer y quinto años.
El análisis de supervivencia en función del estadio al diagnóstico, arrojó los siguientes datos: de los 28 pacientes con sarcomas localizados 14 fallecieron (50%), 2 de ellos durante el primer año, mientras que 14 sobrevivieron (50%); en el grupo de los 10 sarcomas no localizados, encontramos un único superviviente a los 5 años (10%), falleciendo el 40% durante los primeros 12 meses desde el comienzo de la enfermedad; en el grupo de estadio no conocido, 2 pacientes (66,67%) fallecieron en los primeros 5 años y uno (33,33%) permanece vivo. En el subgrupo de localizados fallecidos, calculamos una media de supervivencia de 25 meses (rango 8-67 meses), y en el de no localizados fallecidos de 9,75 meses con un rango 0,75-29 meses.
Con respecto a la supervivencia según localización axial/no axial, distinguimos 13 casos axiales y 28 no axiales. Dentro de los casos axiales, calculamos una tasa de supervivencia del 38,46% a partir del quinto año de seguimiento, del 30,77% entre los años primero y quinto y del 30,77% en los primeros doce meses de seguimiento. En el subgrupo no axial, la mortalidad durante el primer año asciende al 14,29%, un 46,43% sobreviven entre el primer y el quinto año, y un 39,29% siguen vivos al quinto año. Realizamos un subanálisis de los casos axiales diferenciando los casos pélvicos (8 casos, 61,54%) de los extrapélvicos (5 casos, 38,46%), y descubrimos que todos los pacientes con enfermedad pélvica fallecieron antes del final del seguimiento, la mitad de ellos durante el primer año y la otra mitad antes del 2.° año. Todos los casos axiales extrapélvicos, en los que encontramos un tumor epidural, un tumor paravertebral y tres tumores óseos en cuerpos vertebrales de D11, L2 y L3, sobrevivieron al final del seguimiento.
En cuanto a la supervivencia en función de tipo de Ewing, óseo o extraesquelético, encontramos que 13 de los 32 Ewing óseo (40,63%) sobrevivieron a lo largo de los 6 años de seguimiento, 17 (53,13%) fallecieron en los primeros 5 años de enfermedad (4 de los mismos en el primer año
-–12,5%–), y los 2 restantes (6,25%) lo hicieron durante el sexto año de seguimiento. En el grupo extraesquelético, 6 de los 9 (66,67%) fallecieron en los 5 primeros años de seguimiento, 2 de esos 6 durante los doce primeros meses, y tan solo 3 pacientes (33,33%) sobrevivieron al final del seguimiento.
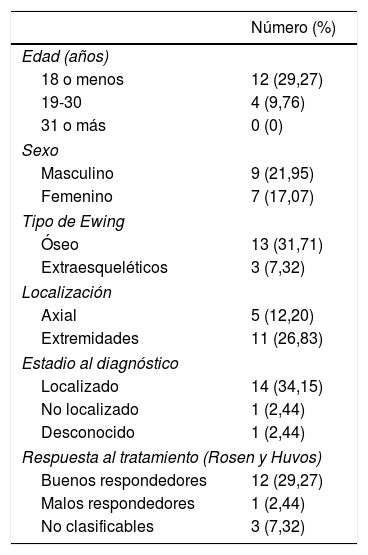
En lo que a tipo de respuesta a terapia neoadyuvante se refiere, 11 de los buenos respondedores (64,71%) sobrevivieron a todo el seguimiento, y los 6 restantes (35,29%) fallecieron no antes de los 14 meses desde el comienzo de la enfermedad. Del grupo mal respondedor, ningún paciente sobrevivió. En la tabla 2 se resumen los datos más relevantes de supervivencia.
Supervivencia a 5 años
| Número (%) | |
|---|---|
| Edad (años) | |
| 18 o menos | 12 (29,27) |
| 19-30 | 4 (9,76) |
| 31 o más | 0 (0) |
| Sexo | |
| Masculino | 9 (21,95) |
| Femenino | 7 (17,07) |
| Tipo de Ewing | |
| Óseo | 13 (31,71) |
| Extraesqueléticos | 3 (7,32) |
| Localización | |
| Axial | 5 (12,20) |
| Extremidades | 11 (26,83) |
| Estadio al diagnóstico | |
| Localizado | 14 (34,15) |
| No localizado | 1 (2,44) |
| Desconocido | 1 (2,44) |
| Respuesta al tratamiento (Rosen y Huvos) | |
| Buenos respondedores | 12 (29,27) |
| Malos respondedores | 1 (2,44) |
| No clasificables | 3 (7,32) |
Del total de sarcomas analizados, 9 resultaron ser metastásicos al diagnóstico (21,95%), 6 óseos –4 en esqueleto axial y 2 en extremidades– y 3 extraesqueléticos –todos en extremidades–. Sometiendo estos datos a un nuevo análisis encontramos que de un total de 13 Ewing asentados sobre esqueleto axial, 4 de ellos (30,78%), correspondían a un estadio IV, mientras que de 28 Ewing con asiento sobre extremidades, 5 de ellos (17,86%) correspondían a un estadio IV al diagnóstico.
Del total de 41 pacientes, 13 evolucionaron hacia la curación de la enfermedad, sin presencia de recidivas locales ni de metástasis durante los 6 años del seguimiento (31,71%). Dos pacientes, 4,88%, sufrieron una recidiva local que fue tratada según el protocolo mencionado anteriormente. Un 21,95% (9 pacientes) desarrollaron metástasis sin recidivas, fundamentalmente óseas y pulmonares, pleurales en un caso y ganglionares en otro caso. Siete pacientes, 17,07%, sufrieron recidivas locales junto con metástasis ganglionares, óseas y pulmonares, encontramos un caso con metástasis hepáticas. Ocho pacientes (19,51%) debutaron con enfermedad metastásica pulmonar y/u ósea, todos evolucionaron al desarrollo temprano de otras metástasis (cerebrales en un caso, pleurales en otro). Un paciente desarrolló un linfoma de células B secundario a la radioterapia adyuvante recibida. En un caso el tumor evolucionó a rabdomiosarcoma en el análisis anatomopatológico de la pieza de resección, desarrollando posteriormente metástasis sistémicas.
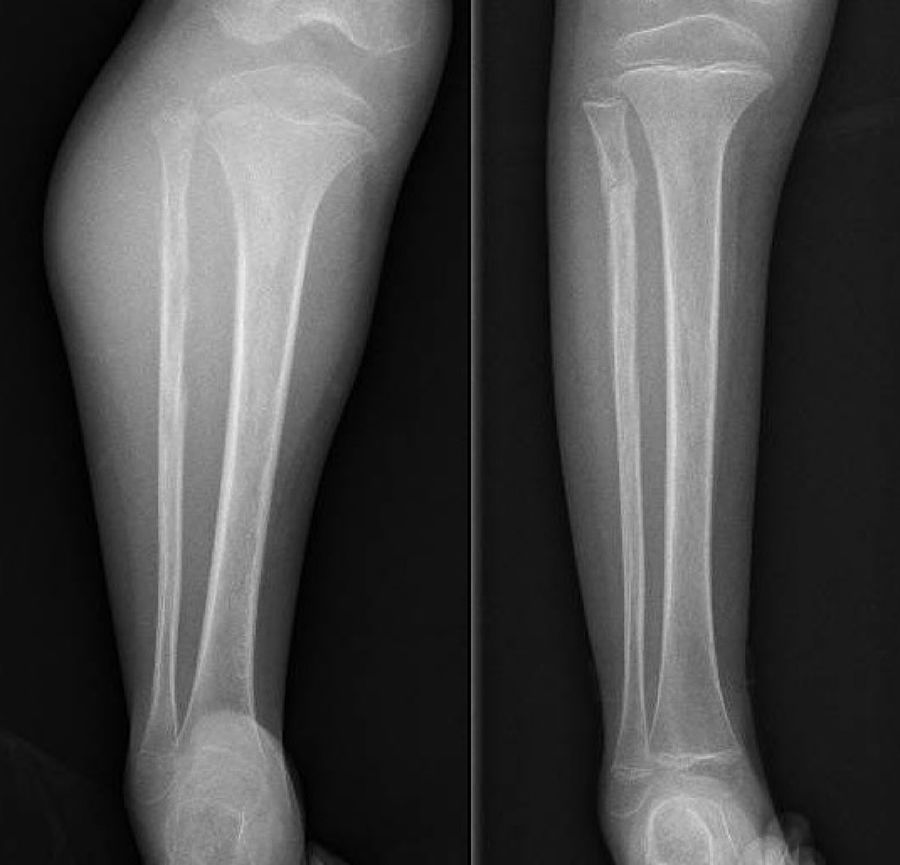
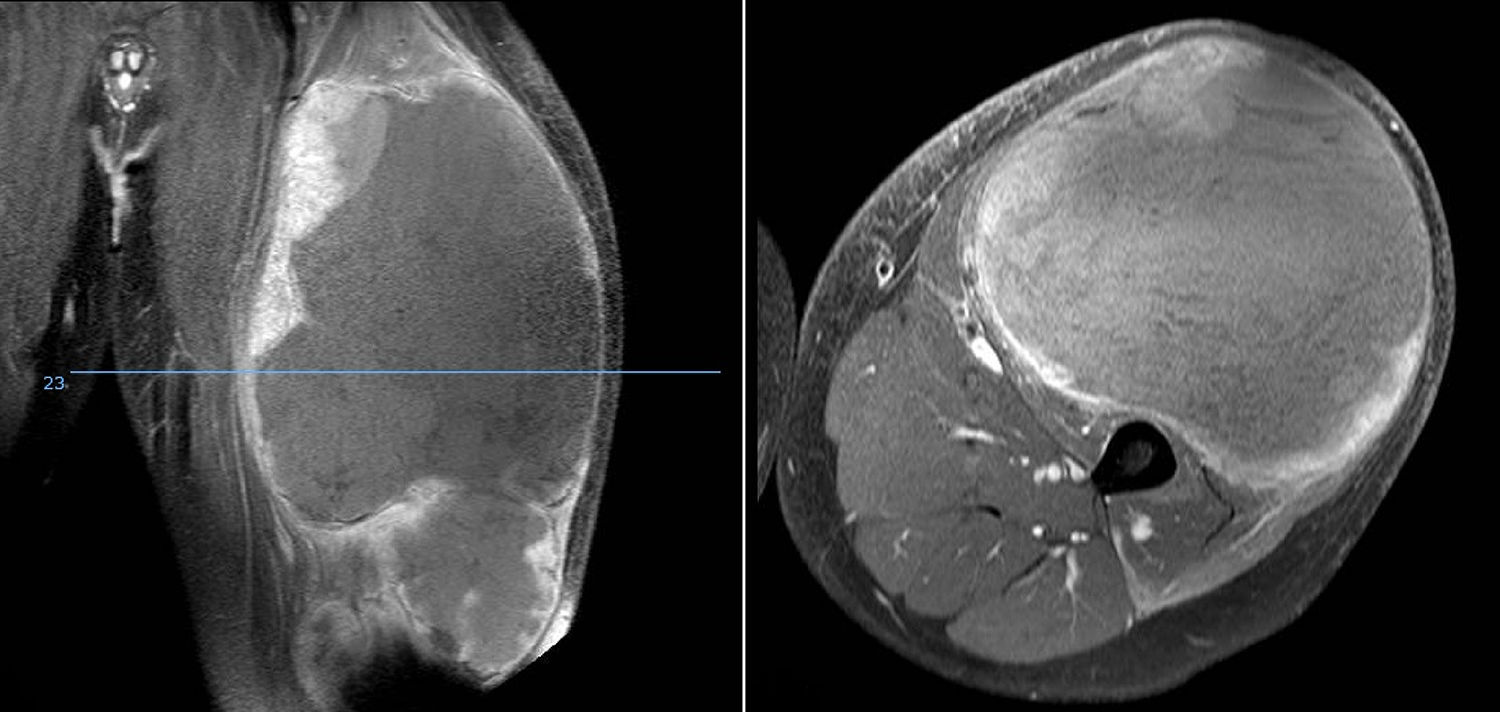
DiscusiónHemos analizado numerosos factores relacionados con el sarcoma de Ewing en una serie de 41 pacientes tratados en un mismo hospital en España durante 6 años. En nuestra serie, 2 tercios de los pacientes fueron varones y un tercio mujeres, siendo casi el 70% menores de 20 años de edad en el momento del diagnóstico anatomopatológico del tumor, datos que corresponden a lo descrito en la literatura1,4,7,8. Raramente se diagnostica en menores de 5 y en mayores de 303,13, en nuestra serie encontramos cinco casos (12,2%) en ese rango de edad: un paciente de 51 años y otro de 49 años de edad con formas extraesqueléticas (muslo –figura 3– y pierna respectivamente) y otros tres pacientes de 31, 32 y 35 años de edad con Ewing óseos en tibia y fémures respectivamente (fig. 4).


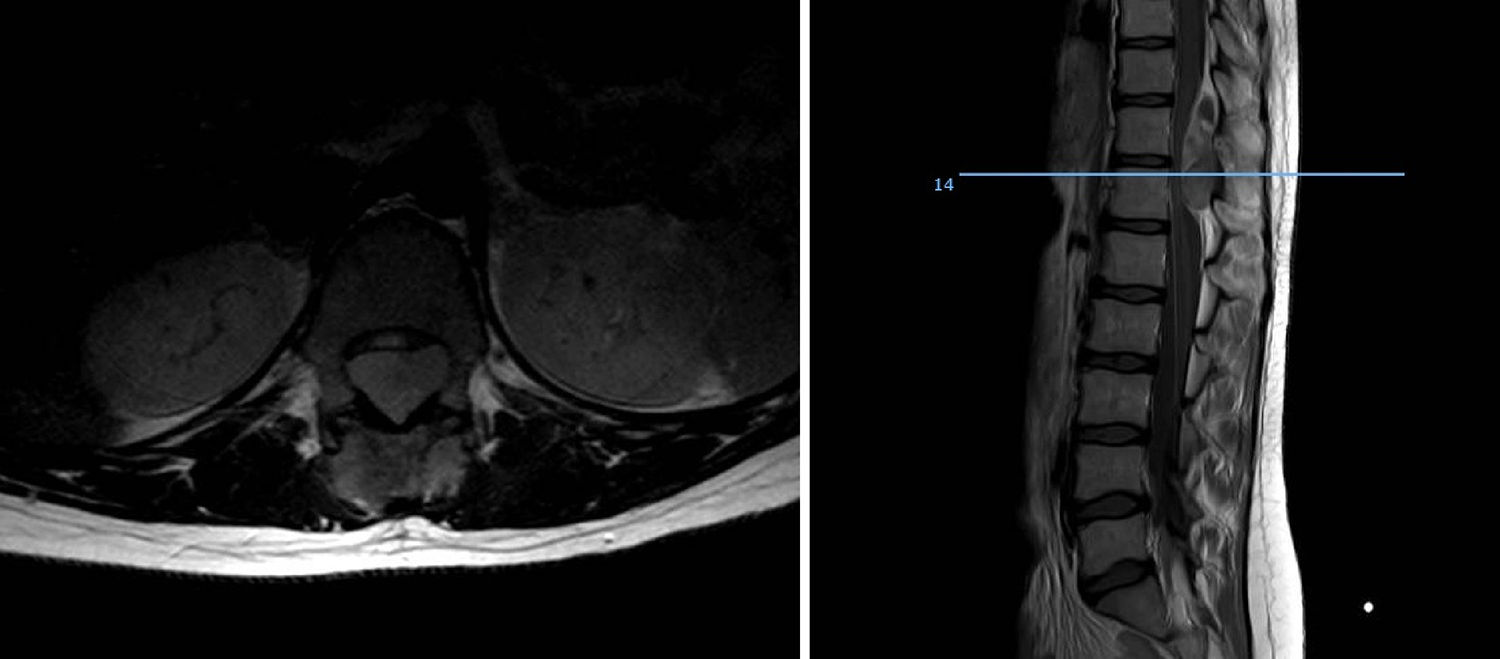
En lo que a localización se refiere, las diáfisis o segmentos metafisodiafisarios de huesos largos, especialmente fémur, constituyen la localización más frecuente de asiento de este sarcoma, en nuestra serie casi la mitad del total de tumores asentaban en dicha localización (18 casos) siendo el fémur el hueso más frecuente, seguido de peroné, tibia y húmero. Los huesos planos se ven afectados con mayor frecuencia que en otros sarcomas óseos en esta enfermedad, un cuarto de nuestros pacientes presentaba asiento en esqueleto axial, 3 en pelvis y 8 en columna vertebral, constituyendo el sacro la sublocalización más común de este grupo. Hasta un 20% son sarcomas extraesqueléticos, tumores de partes blandas sin afectación ósea pero con la misma histología que el Ewing óseo, asentando sobre todo a nivel paravertebral, pared torácica y en miembros inferiores. Nuestro análisis encontró un 21,95% de casos extraesqueléticos los cuales se distribuyeron en miembros inferiores la mayoría y a nivel paravertebral y epidural (2 casos). No encontramos ningún caso de localización epifisaria y solo uno de los pacientes debutó con una fractura patológica, datos que concuerdan con la literatura actual.
Un 21,95% de nuestros pacientes presentó metástasis en el momento del diagnóstico de la enfermedad, dato que se corresponde con el 15-20% descrito en diferentes publicaciones. Casi la mitad de los casos metastásicos al diagnóstico, se trataban de Ewing axiales, ya fueran óseos o extraesqueléticos, encontrando un porcentaje de presencia de metástasis al diagnóstico en Ewing axiales del 30%, en comparación con el 17% que poseían los Ewing emplazados en extremidades. Es decir, en nuestra serie los pacientes con enfermedad axial presentaban casi el doble de posibilidades de corresponder a un estadio IV al diagnóstico que los pacientes con Ewing de localización en extremidades.
Las tasas de supervivencia han aumentado de un 10-15% hasta un 65-70% en los últimos 40 años, mejoría fundamentalmente debida al uso agresivo de la quimioterapia y al tratamiento multidisciplinar4,14,15. Sin embargo, el pronóstico sigue siendo pobre sobre todo en pacientes con enfermedad diseminada o recidiva precoz siendo su supervivencia libre de enfermedad a los 5 años de un 9-30%. En nuestra serie, calculamos una supervivencia global del 85,27% en el primer año y del 43,90% a los 5 años, y una media de supervivencia en los éxitus de 21,95 meses (rango de 0,75-67 meses).
Parece que la edad constituye un factor pronóstico en esta enfermedad, sin embargo sigue existiendo controversia en la literatura6,10,16,17. En nuestro estudio, el análisis de supervivencia según edad destaca que las mejores cifras se obtienen en pacientes pediátricos, un 52,17% del total del subgrupo pediátrico se encontraron vivos a partir del quinto año de seguimiento, un 85% de estos supervivientes contaban con 14 años o menos; con respecto a la serie completa (43,90% de supervivencia a los 5 años) supone que casi el 30 de ese 43,90% de supervivientes son pacientes de 18 años de edad o menos en el momento del diagnóstico. Por otro lado, es en pacientes mayores de 30 años, donde este tipo de tumores son infrecuentes, donde recogimos las peores tasas de supervivencia: no encontramos supervivientes a los 5 años de seguimiento, falleciendo la mitad durante el primer año y la otra mitad entre el primero y el quinto. En el grupo de edad intermedia, encontramos 4 pacientes con una supervivencia mayor de 5 años (33,33%), 2 con supervivencia inferior al año y 6 (50%) cuyo éxitus se produjo entre el primer y quinto años de seguimiento. Se ha postulado que la biología de la enfermedad difiera en función de los distintos grupos de edad, que la enfermedad sea más agresiva en pacientes mayores o que estos respondan peor a los actuales tratamientos10.
Clasificando a nuestros pacientes según su estadio de presentación, la mitad de los sarcomas en estadio localizado sobrevivieron durante todo el seguimiento, mientras que la otra mitad fallecieron a consecuencia de la enfermedad, un 14,28% de ellos durante el primer año, con una media de supervivencia de los fallecidos de 25 meses (rango 8-67 meses). Sin embargo, en el grupo de sarcomas no localizados hallamos un único superviviente a los 5 años (10%), falleciendo el 40% durante el primer año de la enfermedad, con una media de superviviencia de los fallecidos de 9,75 meses. Nuestros datos de supervivencia son inferiores a los reportados en la literatura actual.
La localización pélvica del tumor constituye un factor de mal pronóstico para muchos autores, como para Bacci et al.18 quienes describen en su artículo de 2003 acerca de los Ewing pélvicos no metastásicos que los pacientes afectos de enfermedad pélvica poseen peor pronóstico que aquellos casos extrapélvicos. Plantean que esto pueda ser debido a que la localización pélvica del tumor dificulta el tratamiento local lo cual conlleva altas tasas de recidiva. En nuestro estudio contamos con 13 tumores axiales, 8 de los cuales fueron pélvicos. El 100% de los Ewing pélvicos fallecieron antes del final del seguimiento, la mitad de ellos durante el primer año y la otra mitad entre el primero y el quinto, no llegando la supervivencia en ningún caso a superar los 24 meses. Los casos axiales extrapélvicos presentaron una supervivencia del 100% a partir del quinto año de seguimiento.
Applenbaum et al.19 analizan los Ewing extraesqueléticos en su artículo publicado en la revista Cancer en 2011 y llegan a la conclusión de que estos tumores poseen peor pronóstico en los dos primeros años de la enfermedad a consecuencia de un diagnóstico más tardío y de que aparecen a una media mayor de edad en comparación con los óseos. Aunque nuestro número de casos no nos permite obtener resultados estadísticamente significativos, debemos mencionar que en nuestra serie obtuvimos mejores datos de supervivencia en los Ewing óseos con respecto a los extraesqueléticos (40,63% y 33,33% de supervivencia global respectivamente), y que los dos pacientes de mayor edad de la serie poseían una forma extraesquelética, datos coincidentes con los de la publicación mencionada.
La mayoría de los autores coinciden en que los pacientes que presentan una buena respuesta histológica a la terapia neoadyuvante poseen mejor pronóstico que aquellos malos respondedores1,3,6. Nuestra serie secunda esta afirmación: del total de supervivientes al final del seguimiento –16 pacientes–, 11 pertenecían al subgrupo buen respondedor y los 5 restantes no recibieron terapia neoadyuvante por los motivos previamente descritos, por lo que no pudieron clasificarse según la escala de Rosen y Huvos. No encontramos supervivientes al final del seguimiento entre los malos respondedores.
En lo que a afectación de márgenes se refiere, creemos que no podemos extraer conclusiones de nuestro estudio debido a que un solo paciente correctamente tratado tuvo que ser sometido a una cirugía de ampliación de márgenes y acabó falleciendo a los 15 meses, paciente que correspondía al grupo de malos respondedores. El otro paciente fue intervenido en un centro no de referencia y sin una biopsia previa, siendo derivado a nuestro centro en cuanto la anatomía patológica de la tumoración resecada objetivó la presencia de células pequeñas y redondas para realizar un tratamiento multidisciplinar e individualizado.
Como conclusión, el sarcoma de Ewing es una enfermedad más frecuente en varones y que sigue presentando una alta mortalidad a pesar de los avances médico-quirúrgicos. Por este motivo, algunos autores lo consideran como una enfermedad diseminada desde el inicio, con micrometástasis que no pueden ser detectadas con las técnicas diagnósticas actuales y que son las que realmente condicionan la supervivencia de la enfermedad. La zona metafisodiafisaria femoral es la más afectada, y el sacro el lugar de asiento más común de los axiales. Las formas extraesqueléticas y los pacientes malos respondedores según la escala de Rosen y Huvos presentan una tasa de mortalidad no solo superior sino también precoz con respecto a los demás. Los Ewing pélvicos en general presentan un diagnóstico tardío lo cual les aporta mayor probabilidad de ser metastásicos en el momento del diagnóstico, característica que ensombrece el pronóstico de los mismos. Las mejores herramientas con las que contamos en la actualidad para ofrecer a los pacientes las máximas probabilidades de curación son el diagnóstico precoz y la terapia multidisciplinar en centros de referencia.
Nivel de evidenciaNivel de evidencia IV.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
