




El linfoma primario del sistema nervioso central (LPSNC) es una enfermedad poco frecuente y agresiva, forma de linfoma no-Hodgkin (LNH) en pacientes inmunocompetentes que representa el 4% de todas las neoplasias intracerebrales y un 1-2% de los linfomas extranodales1. Las condiciones asociadas a inmunodepresión aumentan el riesgo de padecer un LPSNC2,3.
Comunicamos un caso de LPSNC de células B en una paciente inmunocompetente. Paciente de 80 años con antecedentes de asma, HTA parcialmente dependiente para las actividades básicas de la vida diaria (índice de Barthel previo al ingreso de 80), que ingresó en la unidad de convalecencia del Hospital Corporació Sanitaria Parc Tauli por artroplastia total de cadera izquierda. Durante el ingreso presentó declive funcional, lenta recuperación de la marcha y se detectó también alteración la memoria reciente e inatención. En exploración física neurológica destacaba desorientación parcial en tiempo y espacio, orientada en persona, déficits a nivel de funciones ejecutivas con inatención importante con déficits en la memoria de trabajo y la reciente, apraxias visual-espaciales e ideo motoras, pero sin alteración de la nominación ni de la comprensión. Sin otros déficits neurológicos asociados. Cuestionario de Pfeiffer de 6 errores y test de Mini-Mental de 17.
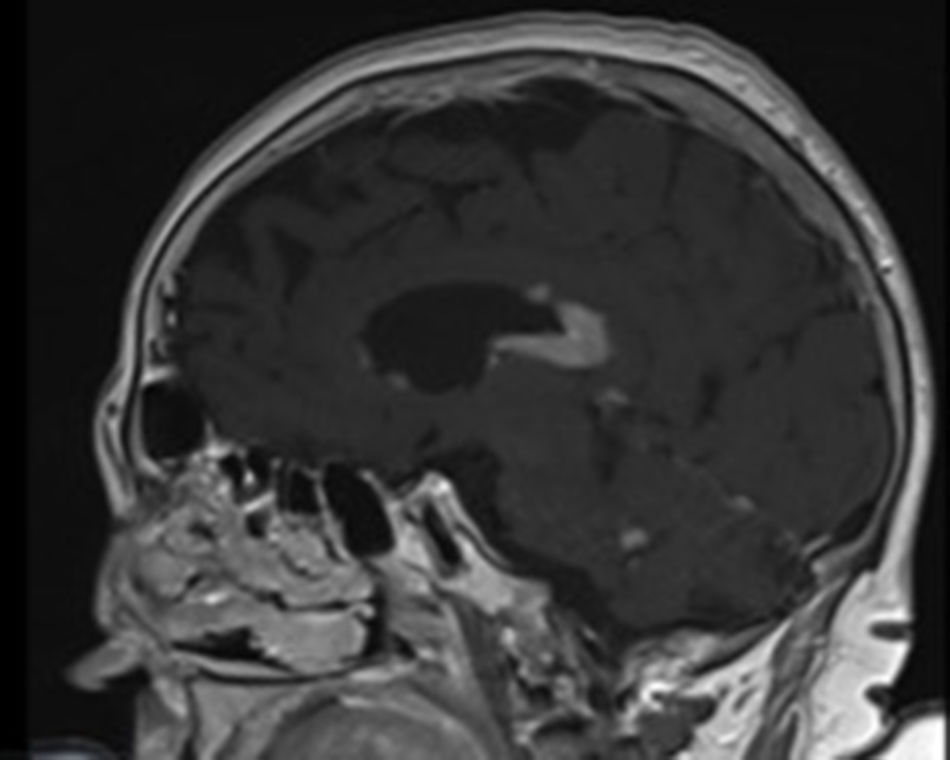
Se realizó análisis sanguíneo con hemograma, función renal, hepática y tiroidea, proteinograma, vitamina B12, ácido fólico, LDH y serologías VIH y sífilis que resultó dentro de la normalidad salvo la presencia de anemia normocítica y normocrómica con Hb 10g/l (120-160). Se realizó TC craneal que objetivó una lesión infiltrativa a nivel cuerpo calloso. Posteriormente se realizó RMN cerebral que mostraba un proceso tumoral a nivel del cuerpo calloso y periependimario, compatible con proceso linfoproliferativo con atrofia cerebral y leuco encefalopatía hipóxica (fig. 1). Se determinó a nivel plasmático ß2-microglobulina 3,3μg/ml (0,8-2,2) que resultó elevada y también inmunofenotipo de sangre periférica que resultó dentro de la normalidad. Se completó estudio de extensión con TC torácica-abdominal que no mostraba enfermedad en otros niveles ni adenopatías. Se valoró por el servicio de neurología y se realizó punción lumbar de la que resultó líquido cefalorraquídeo (LCR) amarillento, a nivel de bioquímica destacaba proteinorraquia de 2g. Histológicamente el LCR no mostró células malignas ni población monoclonal, pero con moderada cantidad de linfocitos de fenotipo T. Se realizó citometría de flujo de LCR en la que se detectó antígenos CD3, CD454, FMC7, CD20, CD22, CD19, CD79b positivos y una población linfoide B policlonal con un predominio de linfocitos T.

También se cultivó LCR para micobacterias y bacterias que resultó negativo. Delante del diagnóstico de LPSNC, se hizo valoración multidisciplinar (geriatría, neurología, hematología y cuidados paliativos) y finalmente se decidió realizar tratamiento conservador y sintomático con seguimiento en domicilio de la paciente por la unidad de cuidados paliativos de nuestro centro.
La paciente a los 3 meses ingresó por somnolencia secundaria a progresión de la enfermedad con desenlace fatal.
El LPSNC es un LNH agresivo. Se origina en cerebro, ojos, leptomeninges o médula espinal, sin evidencia de linfoma sistémico en el momento del diagnóstico4. Su máxima incidencia se sitúa en la sexta década de la vida. Su incidencia se ha duplicado en las últimas 2 décadas, constituyendo un 4% de los tumores primarios del sistema nervioso central5,6.
La forma clínica más frecuente es aparición de focalidad neurológica, seguida de síntomas neurológicos-psiquiátricos, crisis epilépticas y síntomas oculares. Las lesiones son solitarias en el 65% de los casos, sus localizaciones más frecuentes son: hemisferios 38%, tálamo/ganglios basales 16%, cuerpo calloso 14% y región periventricular 12%1. Se completa estudio con TAC torácica-abdominal, punción lumbar para citología y citometría de flujo y analítica con LDH y ß2-microglobulina. Desde el punto de vista histológico-inmunofenotípico la mayoría expresan Ag CD20 y CD797.
Se describen 5 factores que comportan peor pronóstico: >60 años, LDH elevado, hipeproteinorraquia, afectación de estructuras cerebrales profundas periventriculares y ECOG performance status ≥2. Si no hay factores de riesgo, el 80% de pacientes sobreviven a los 2 años, con 2 o 3 lo hace un 48% y con 4 o 5 factores solo un 15% sobreviven a los 2 años8. La supervivencia media sin tratamiento es de 2-4 meses tras el diagnóstico.
El tratamiento más descrito en la las publicaciones es radioterapia y quimioterapia en los que se han descrito diferentes esquemas con metotrexato intratecal, rituximab y otros agentes quimioterápicos, la cirugía solo se realiza si su localización lo permite9,10.
En este caso se conjugan varios factores que lo hacen singular como son el la edad y la forma de presentación clínica, y posteriormente una evolución rápida y fatal.
