




En los últimos años, gracias a la incorporación de la gammagrafía con 99mTc-ácido 3,3-difosfono-1,2-propanodicarboxílico (99mTc-DPD) en el diagnóstico no invasivo1, y ya más recientemente con la posibilidad de tratamiento tras los resultados del ensayo Transthyretin Amyloidosis Cardiomyopathy Clinical Trial2, se ha incrementado de forma exponencial el interés por una entidad, la amiloidosis asociada a la transtiretina (ATTR) que, pese a su presumiblemente elevada prevalencia en su forma salvaje o wild-type, había tenido, hasta la fecha, un rol residual en el campo de la insuficiencia cardíaca (IC).
La transtiretina (TTR) o prealbúmina es una proteína plasmática, tetramérica, de síntesis predominantemente hepática, encargada del transporte de la tiroxina y del retinol3. Su forma nativa, tetramérica, puede disociarse en dímeros y monómeros, los cuales pueden plegarse y ensamblarse en fibras (fibrillas de amiloide) y depositarse en diferentes órganos y tejidos, dando lugar a la amiloidosis4.
Hasta la fecha se conocen 2 tipos de ATTR según la existencia o no de mutación en el gen que codifica la TTR: la forma hereditaria o familiar, o más recientemente llamada variante (ATTRv), y la forma natural o salvaje, o más comúnmente conocida como wild-type (ATTRwt)5. Mientras la ATTRv sigue siendo considerada una enfermedad rara, con una baja prevalencia (inferior a 1 por cada 100.000 habitantes)6, la ATTRwt (erróneamente llamada «amiloidosis senil», término que afortunadamente en la actualidad está o debería estar completamente en desuso) tiene una prevalencia desconocida debido, en gran parte, a su infradiagnóstico6. No obstante, dada la evidencia objetivada, tal y como publicaron Maurer et al.7, probablemente se trate de la amiloidosis cardíaca más frecuente, hecho que le confiere una relevancia clínica que no había tenido hasta la actualidad. Con todo ello, creemos que los casos que hemos podido detectar a día de hoy solo configuran la cumbre de un iceberg de una magnitud difícil de imaginar.
Clásicamente se ha asociado la ATTRwt con la población masculina (alrededor del 90%) y de edad avanzada (edad media del diagnóstico alrededor de 76 años)8 sobre todo, en gran medida, por un diagnóstico tardío. No obstante el paradigma de la enfermedad está permutando hacia una nueva realidad, con una prevalencia creciente en mujeres, de más del 50% en algunas cohortes9 y con la documentación de algún caso de ATTRwt incluso en pacientes menores de 50 años de edad10, por lo que debemos evitar pensar en la ATTRwt únicamente en la población de edad avanzada masculina y plantear su diagnóstico a un abanico más amplio de pacientes. Un interesante estudio español evaluó pacientes de más de 60 años ingresados por IC y fracción de eyección preservada e hipertrofia ventricular izquierda (HVI≥12mm) demostrando una prevalencia del 13% de ATTRwt, dándonos una idea de la relevancia de esta enfermedad en este subgrupo de pacientes con IC y fracción de eyección preservada9.
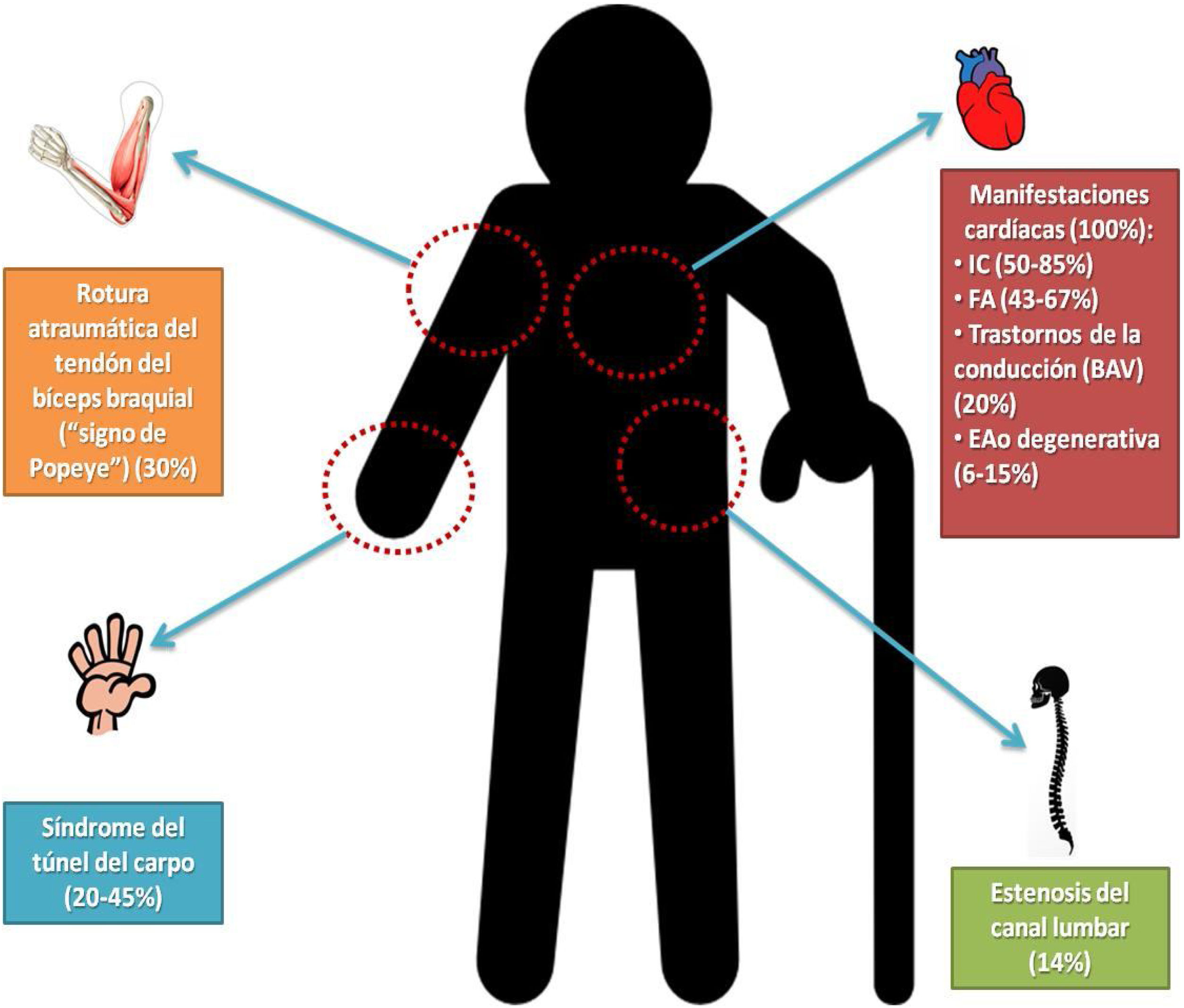
Clínicamente (fig. 1) la ATTRwt presenta manifestaciones cardíacas de forma constante (en el 100% de los casos), derivadas de la infiltración del miocardio por parte de las fibras de amiloide (típicamente a nivel septal), del tejido de conducción y/o del aparato valvular11. Tal y como describen González-López et al.11 en su revisión, la forma más frecuente de presentación cardíaca de la ATTRwt es la IC (50-85%), seguida de la presencia de arritmias auriculares (principalmente fibrilación auricular [FA]) (43-67%)11,12 o afectación del nodo sinusal o el nodo auriculoventricular (AV), que conllevará la presencia de bloqueos AV (BAV) (alrededor de un 20%)13, y entre el 6% y el 15% de los pacientes pueden presentar valvulopatía aórtica en forma de estenosis aórtica degenerativa (normalmente con bajo flujo-bajo gradiente)14–16. En muchas ocasiones la infiltración miocárdica por fibras de amiloide puede mimetizar otras formas de miocardiopatía hipertrófica o infiltrativa7, pudiendo conllevar, en algunas ocasiones, errores diagnósticos. Según algunas series, como la que describen en su artículo Lousada et al.17, más del 50% de los pacientes ATTRv y el 39% de los ATTRwt recibieron un diagnóstico erróneo; de ellos el 76% y el 75%, respectivamente, recibieron tratamiento para ello. Por lo tanto, la correcta fenotipificación de los pacientes con IC y la identificación de aquellos casos susceptibles de presentar ATTR deben ser un reto y una motivación para todos aquellos profesionales que atienden a pacientes con IC, como los geriatras.

Por otra parte, a nivel extracardíaco, a diferencia de lo que sucede en la ATTRv, en la que las manifestaciones extracardíacas son más prevalentes y heterogéneas, en la ATTRwt dichas manifestaciones se suelen limitar a la presencia de síndrome del túnel del carpo (sobre todo bilateral), que es la manifestación extracardíaca más prevalente (entre el 20% y el 45%)7,18, de estenosis del canal lumbar19 o de rotura atraumática del tendón del bíceps o «signo de Popeye», todas ellas derivadas del depósito de las propias fibrillas de amiloide11. Pese a que su aparición no es constante en los pacientes con ATTRwt, el hecho de detectar manifestaciones extracardíacas en pacientes sin diagnóstico de ATTR pueden ser consideradas como signos de alarma, y motivar a incluir la enfermedad en el diagnóstico diferencial durante el estudio etiológico de estas entidades promoviendo, así, un diagnóstico precoz.
La aparición de síndrome del túnel del carpo suele ser más precoz que la afectación cardíaca, llegando a preceder su aparición en varios años (de 5 a 10 años del diagnóstico de la miocardiopatía)18,20 y, además, su diagnóstico per se puede asociarse a un incremento del riesgo de un futuro diagnóstico de amiloidosis cardíaca, IC u otros eventos adversos como fibrilación auricular, bloqueo auriculoventricular o la necesidad de implante de marcapasos21. Además, según los resultados del estudio de Milandri et al.18, debe considerarse como un marcador de mal pronóstico en aquellos pacientes con ATTR, sobre todo en la forma wild-type, independientemente de la afectación cardíaca. Por lo tanto, se trata de la principal manifestación extracardíaca a considerar por su elevada prevalencia y su relevancia pronóstica en el curso de la enfermedad.
El motivo principal que conlleva un diagnóstico tardío, cuando la enfermedad se encuentra ya en un estadio avanzado de su curso natural, es la escasa detección o el bajo índice de sospecha en aquellos estadios iniciales pauci-sintomáticos22. El diagnóstico precoz de la miocardiopatía por ATTR es clave debido a que el pronóstico de estos pacientes decae de forma progresiva debido al continuo depósito de amiloide y a la disfunción orgánica derivada7. Por lo tanto, tal y como argumentan Maurer et al.7 y Emdin et al.23, debemos sospechar la presencia de miocardiopatía por ATTR en aquellos pacientes con IC, síncope o bradiarritmia, junto con hallazgos sugestivos de amiloidosis cardíaca en las pruebas de imagen (engrosamiento del ventrículo izquierdo no explicado por historia clínica de hipertensión arterial, especialmente a nivel septal, con voltajes QRS normales o reducidos).
Uno de los elementos clave que ha permitido una detección mayor y más precoz de los pacientes con ATTR ha sido la incorporación de la gammagrafía con 99mTc-DPD, gracias a la cual es posible un diagnóstico no invasivo de la enfermedad, lo que facilita la accesibilidad al diagnóstico a la mayor parte de los profesionales sanitarios involucrados en este ámbito. La combinación de la presencia de signos clásicos de amiloidosis cardíaca por técnicas de imagen, un grado 2-3 de captación miocárdica en gammagrafía 99mTc-DPD/PYP (99mTc-pirofosfato), junto con la exclusión de una proteína monoclonal (con el fin de descartar AL) en sangre y en orina, confiere una sensibilidad y un valor predictivo positivo del 100% para el diagnóstico de ATTR1.
En lo que se refiere a tratamientos modificadores del curso de la enfermedad, hasta hace poco las opciones terapéuticas se limitaban al trasplante cardíaco y/o hepático, pero tras los resultados obtenidos con el uso de tafamidis en pacientes con miocardiopatía por ATTR2, y otros resultados preliminares, especialmente en lo que atañe a la supresión de la expresión del gen de la TTR, como los obtenidos con patisiran24 e inotersen25, pese a que no se han evaluado específicamente en pacientes con miocardiopatía por ATTRwt, abren un futuro esperanzador en el tratamiento de esta enfermedad.
Con todo ello, la ATTR se está convirtiendo en una enfermedad cada vez más prevalente a medida que incrementa su diagnóstico, especialmente el de la ATTRwt, de la cual empezamos a vislumbrar indicios de este iceberg que desconocíamos. Estos datos hacen sospechar que el futuro impacto de la ATTR, sobre todo la wild-type, en los pacientes con IC puede ser muy elevado, hecho que debe motivar a todos los profesionales a optimizar los procesos de fenotipado de todos los pacientes con IC, con el fin de poder ofrecer tratamientos dirigidos y efectivos que pueden conllevar cambios significativos en el curso y pronóstico de la enfermedad.
