




La amiloidosis primaria es un proceso debido al depósito de sustancia proteinácea insoluble tipo AL (cadenas ligeras de inmunoglobulinas monoclonales), que se asocia en ocasiones a mieloma múltiple. Por lo general, produce afectación multisistémica. Las manifestaciones cardiacas pueden estar ausentes o ser relativamente menores, y más raramente constituyen la única manifestación de la enfermedad1. La miocardiopatía amiloidea se presenta en forma de insuficiencia cardiaca por la restricción del miocardio, y tiene un mal pronóstico por la rápida y fatal evolución del cuadro. Su incidencia es 0,9 por 100.000 personas-año y afecta por igual a ambos sexos, habitualmente entre 50 y 70 años2. La supervivencia media es de 13 meses aproximadamente desde el diagnóstico, siendo el compromiso de la función cardiaca y renal por afectación restrictiva-infiltrativa la responsable del mal pronóstico3.
Presentamos un caso de amiloidosis primaria con afectación y expresividad clínica exclusivamente cardiaca en una paciente con disnea progresiva y síntomas constitucionales.
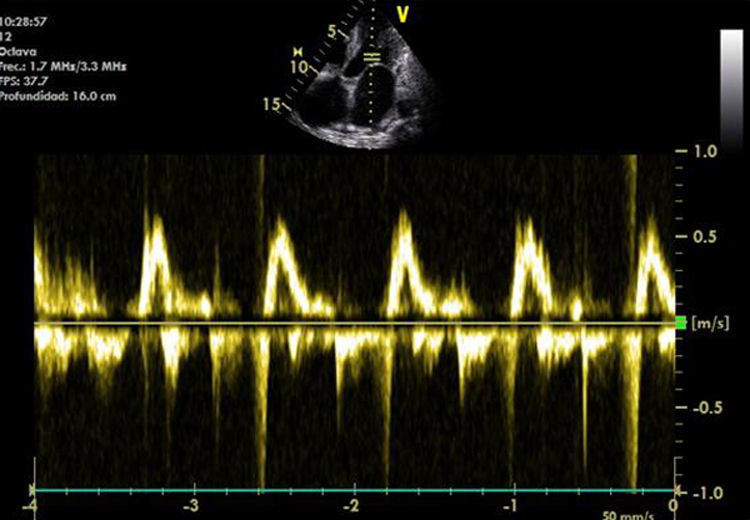
Mujer de 74 años, sin antecedentes previos de interés, que presenta síntomas constitucionales, disnea a moderados esfuerzos, ortopnea e hinchazón de extremidades inferiores de 18 meses de evolución. Valoración geriátrica integral: a) escala de Lawton-Brody para actividades instrumentales: 7; b) test de Pfeiffer: 0; c) escala de Yasevage: 6; d) escala de Gijón: 9. En la exploración destacaba tendencia a la hipotensión; ingurgitación yugular a 30°. En la auscultación cardiopumonar presentaba tonos cardiacos muy apagados y crepitantes en ambos campos pulmonares inferiores. El abdomen no presentaba anomalías. Tenía edemas con fóvea en ambos miembros inferiores. El electrocardiograma mostraba ritmo sinusal, aunque los voltajes eran bajos. La radiografía simple de tórax mostraba cardiomegalia global. Se realizó un ecocardiograma donde se observaba un ventrículo izquierdo con hipertrofia moderada, dilatación biauricular, presión arterial pulmonar estimada de 45-50mmHg y una fracción de eyección del 60%, compatible con miocardiopatía restrictiva-infiltrativa (fig. 1). Se instauró tratamiento con diuréticos y se realizó estudio para detección de enfermedad sistémica. En el estudio analítico, la hematimetría y la coagulación fueron normales; la velocidad de sedimentación globular fue de 54mm la primera hora. El estudio bioquímico general que incluía parámetros de función renal, perfil lipídico y hepático, así como las hormonas tiroideas fueron normales. Los ANA, ANCA y factor reumatoide fueron negativos y la determinación del complemento estaba dentro de la normalidad. El proteinograma mostraba hipergammaglobulinemia con determinación de IgG de 2.460mg/dl (639-1.349mg/dl), IgA de 194mg/dl (70-132mg/dl), y la de IgM fue normal. Asimismo, la determinación de las cadenas ligeras kappa fue de 56,4mg/l (3,3-19,4mg/l), y lambda de 135mg/l (5,7-26,3 md/l). La proteinuria de Bence-Jones fue negativa. La ecografía abdominal mostraba un hígado de estasis, sin otros hallazgos. La radiografía de cráneo no presentaba lesiones líticas. Se obtuvo muestra de grasa abdominal por biopsia que no mostraba hallazgos histológicos patológicos. Posteriormente, se realizaron biopsia rectal y de médula ósea apreciándose en ambas depósito amiloide con tinción rojo Congo e inmunohistoquímica negativa a cadenas ligeras. Con el diagnóstico de amiloidosis sistémica primaria se instauró tratamiento con melfalán y prednisona. La paciente presentó en los primeros meses mejoría de su clínica cardiológica, aunque con un temprano y progresivo deterioro que obligó al empleo de fármacos diuréticos a dosis plenas con escasa respuesta. Se propuso para trasplante cardiaco al hospital de referencia desestimándose por la rápida progresión y el mal estado funcional de la paciente. A los 7 meses del diagnóstico se produce el fallecimiento de la paciente en situación de insuficiencia cardiaca refractaria.

La amiloidosis AL o primaria es una discrasia de células plasmáticas que suele provocar infiltración multiorgánica afectándose el corazón en más del 50% de los casos. No obstante, la expresión clínica exclusivamente cardiaca sin manifestaciones de otros órganos es una forma de presentación muy infrecuente1,4. Menos del 5% de las amiloidosis primarias tienen manifestaciones clínicas de índole cardiológica de forma aislada; sin embargo, la sensibilidad diagnóstica de las biopsias de otros órganos suele ser menor del 80%5. El tratamiento sintomático de la miocardiopatía amiloidea basado en fármacos diuréticos está dificultado por la tendencia a la hipotensión arterial de los pacientes. Los inhibidores de la enzima de conversión de la angiotensina y los betabloqueantes son mal tolerados, y los antagonistas del calcio y la digoxina no se aconsejan (tendencia a unirse a la proteína anómala)6. El manejo terapéutico actual de la amiloidosis (quimioterápicos y/o trasplante de médula ósea) tiene una eficacia muy discreta7. De forma general, en pacientes con miocardiopatía muy avanzada, se desaconseja el trasplante de médula ósea. Por otro lado, la indicación de trasplante cardiaco es muy controvertida por la afectación multiorgánica y la probabilidad de recidiva sobre el órgano trasplantado7,8. En casos seleccionados, la estrategia más aceptada sería realizar primero el trasplante cardiaco (de alto riesgo) y después el tratamiento etiológico de la amiloidosis (quimioterapia y/o trasplante de MO)7.
La formación de equipos multidisciplinarios que permitiesen un tratamiento integral que incluya trasplante multiorgánico, junto a la aparición de tratamientos más efectivos y nuevas técnicas para el diagnóstico y monitorización del tratamiento, contribuirían sin duda a mejorar el pronóstico de esta enfermedad8.
