




Un hombre de 81 años fue ingresado por un cuadro de 4 meses de astenia, anorexia, pérdida de 8kg de peso, vértigo y ataxia de la marcha de instauración subaguda.
Presentaba antecedentes de hipertensión arterial, dislipidemia, fibrilación auricular en tratamiento con warfarina y un infarto agudo de miocardio inferior, así como ligera anemia por bloqueo del hierro de 2 años de duración.
Destacaba nistagmo horizontorrotatorio de la mirada lateral a la izquierda. La marcha era inestable, con lateralización a la derecha. El Barthel era 45 y el Pfeiffer 0. El resto de la exploración era normal salvo tonos cardíacos arrítmicos. Presentaba Hb 110g/l, Hto% 0,34, VCM 81 fL, VSG 54mm/h y un INR 2,45, discreta elevación policlonal de alfa-2-globulinas (12,1%) y de gammaglobulinas (19,8%). El resto de la analítica era normal. La radiografía de tórax mostraba cardiomegalia y en el electrocardiograma se observaba fibrilación auricular. Se realizó una TC toracoabdominal, sin hallazgos relevantes.
Se orientó el diagnóstico como vértigo de origen central y se solicitó una resonancia magnética nuclear (RMN) craneal, realizada sin difusión, que objetivó una lesión isquémica aguda en hemisferio cerebeloso izquierdo y áreas de desmielinización de origen isquémico crónico periventriculares. Se realizó un ecocardiograma que mostró hipocinesia posteroinferobasal sin trombo y una ecografía Doppler de troncos supraaórticos que mostró estenosis de 1-15% de arterias carótidas internas. Las arterias subclavias y vertebrales eran normales.
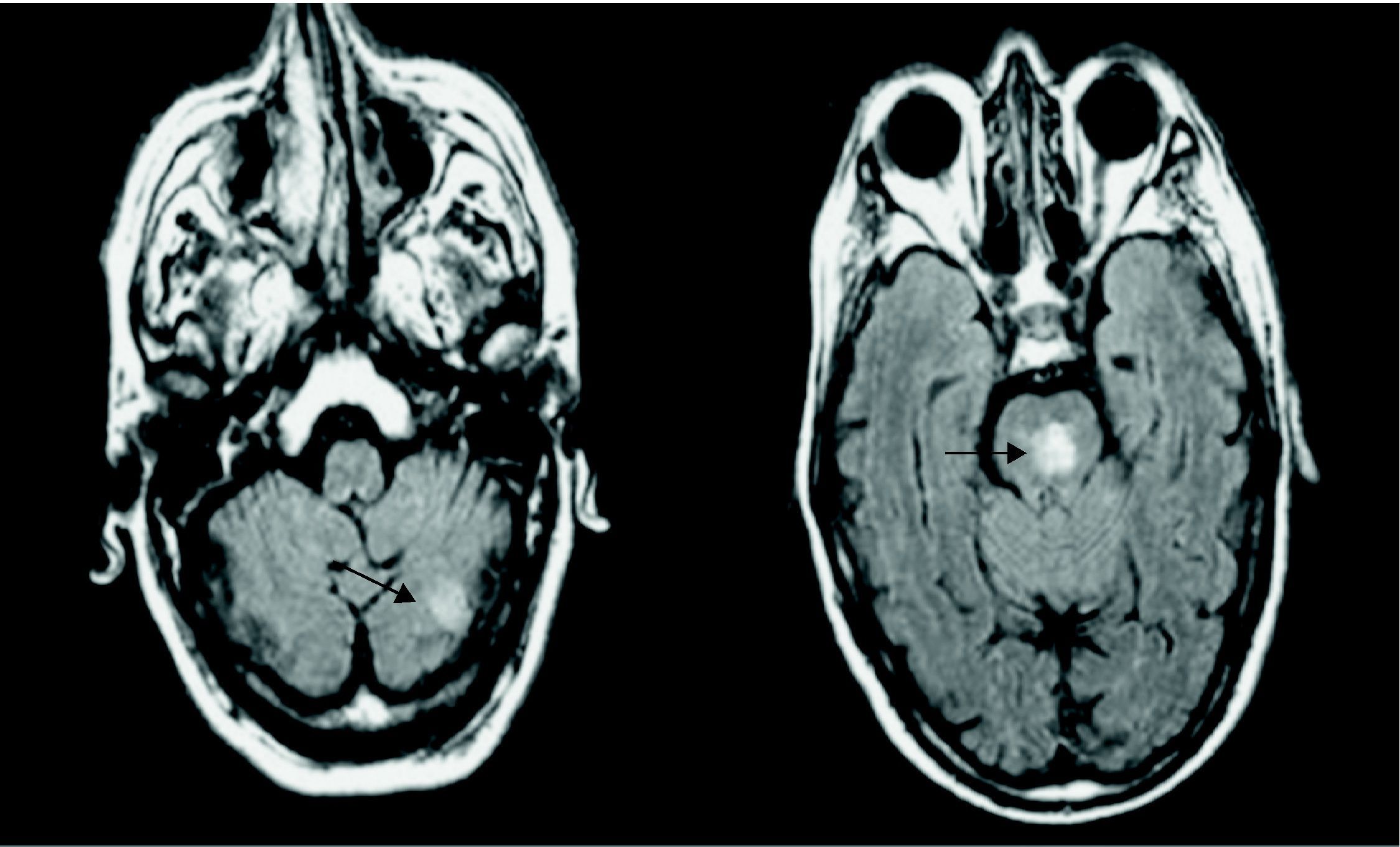
Una nueva RMN a los 10 días (fig. 1), esta vez con difusión, reveló isquemia crónica de sustancia blanca supratentorial, infarto subagudo en hemisferio cerebeloso izquierdo y un nuevo infarto agudo protuberancial izquierdo no presente en el examen previo.

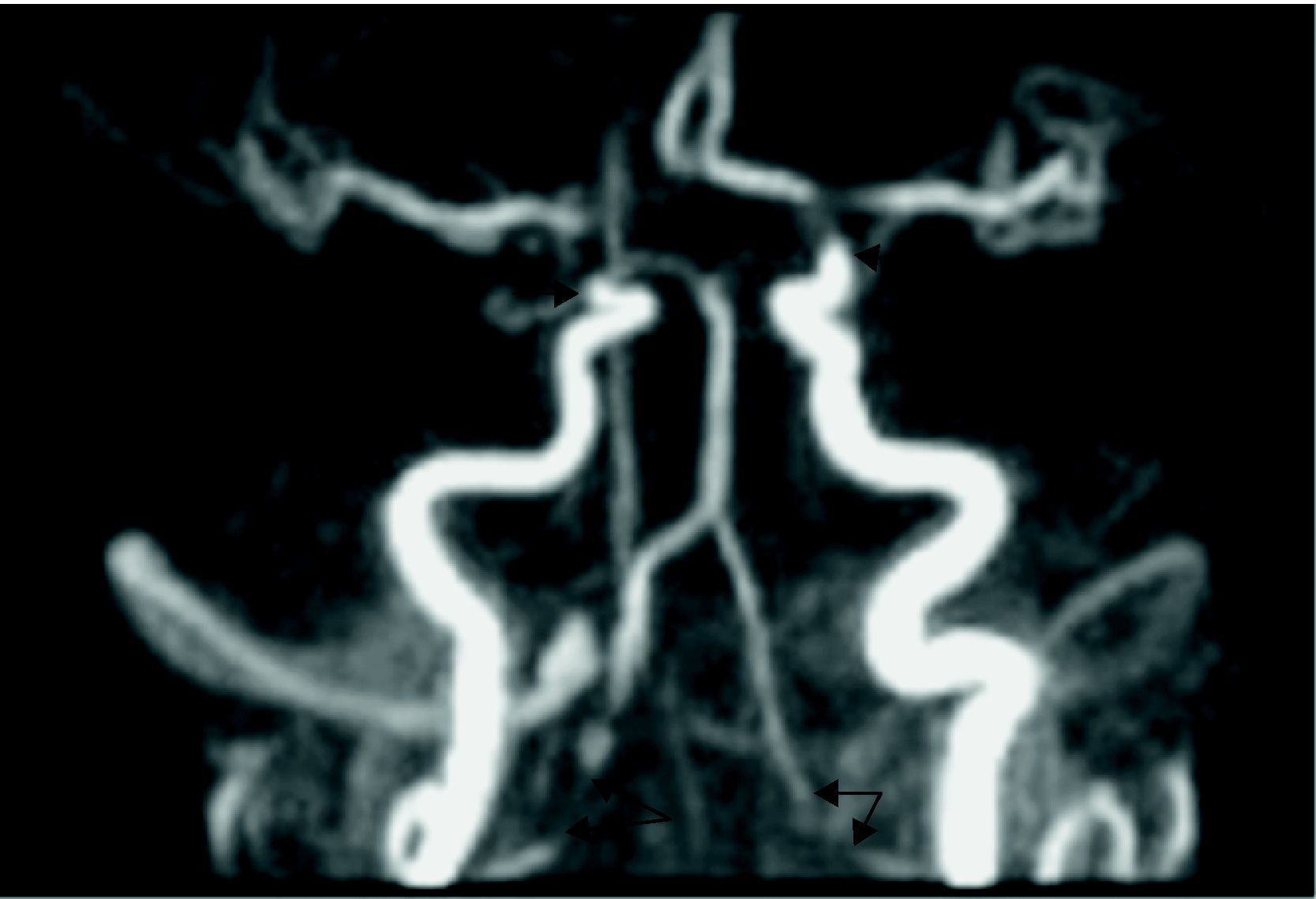
Apareció en ese momento de la evolución un cuadro de debilidad y algias de cintura escapular, por lo que se sospechó una polimialgia reumática asociada a una arteritis de células gigantes pese a que no existía cefalea y las arterias temporales eran normales en la exploración física. Se instauró tratamiento con prednisona a dosis de 1mg/kg/día y se solicitó una angio-RMN (fig. 2) que objetivó flujo filiforme en la carótida interna izquierda intracavernosa por estenosis severa y oclusión de sifón carotídeo derecho. Ambas arterias vertebrales presentaban severa estenosis al inicio de su porción intradural. Finalmente, se realizó biopsia de la arteria temporal cuyo resultado fue compatible con arteritis de células gigantes.

En los días que siguieron al inicio de la corticoterapia mejoraron claramente la astenia, la anorexia y la debilidad muscular. La ataxia y el vértigo central habían mejorado parcialmente. En el control ambulatorio un mes más tarde el paciente era capaz de caminar sin ayuda, aunque todavía refería cierta inestabilidad. Sin embargo, presentaba claros signos de deterioro cognitivo con moderada apraxia. El Barthel era de 35 y el Pfeiffer con 4 errores. Al año de seguimiento, el paciente camina solo sin ayuda y presenta únicamente un leve deterioro cognitivo que le permite mantener una conversación, sin necesitar ayuda para vestirse o lavarse. El Barthel actual es de 65 y en el Pfeiffer presenta 2 errores.
La angio-RMN repetida a los 2 meses del diagnóstico no demostró reperfusión de los troncos supraaórticos a pesar del tratamiento y de la buena evolución clínica.
La arteritis de células gigantes (ACG) es una enfermedad de naturaleza autoinmune que afecta a la lámina elástica de las capas media y adventicia de las arterias de mediano y gran calibre, principalmente de la cabeza y el cuello. La afectación neurológica fue descrita por Caselli et al en 1988 en forma de síndromes neurooftalmológicos, neuropatía, accidente vascular cerebral (AVC) o accidente isquémico transitorio (AIT), síndromes neurootológicos, temblor, síndromes neuropsiquiátricos, entumecimiento de la lengua y mielopatía1. La incidencia de episodios isquémicos vasculares cerebrales se ha estimado en 3-4% en este grupo de pacientes, debidos a estenosis u oclusión extradural vertebral o carotídea más que a una afectación vascular intradural2. Este hecho se debe a la pérdida por parte de las arterias de la capa elástica una vez atraviesan la duramadre2. En la población general se ha descrito un ratio de 5:1 entre la afectación vascular del territorio carotídeo frente al territorio vertebrobasilar. En cambio, en los pacientes con ACG esta ratio resulta ser de 3:23, e incluso se invierte si nos ceñimos al período que cursa entre el inicio de los síntomas y el primer mes que sigue al inicio del tratamiento, especulándose que este hecho podría estar en relación con el menor calibre de los vasos vertebrobasilares4. El desarrolo de AVC debido a afectación extracraneal se debe, por este orden, bien a oclusión arterial por la propia vasculitis, a un émbolo arteria-arteria o bien a progresión del trombo2. En contraposición, la afectación intracraneal es menos frecuente y casi siempre extradural por los motivos antes mencionados. De hecho, la afectación intradural se debe en la mayor parte de los casos a un episodio embólico arteria-arteria más que a un proceso arterítico en dicha localización5. Muy infrecuentemente existe afetación intracraneal e intradural aislada y, en ese caso, se debería diferenciar con una vasculitis primaria del sistema nervioso central2,6.
Muy pocos son los casos descritos en la literatura médica que se hubieran manifestado exclusivamente en forma de AVC como el caso que aquí presentamos7,8. En algunos casos también se ha descrito un inicio insidioso de los síntomas neurológicos y no agudo como se esperaría en la afectación vascular cerebral9.
En los pacientes con ACG y afectación vascular cerebral se ha constatado un menor grado de anemia normocítica, que incluso en caso de estar presente podría actuar de factor protector frente a la afectación neurológica4,10. Se ha postulado revisando muestras de autopsias la posibilidad de que el propio proceso inflamatorio induzca la neoangiogénesis, protegiendo así de accidentes isquémicos cerebrales11. Por tanto, los pacientes con menor grado de anemia y menor repercusión en los reactantes de fase aguda comprenderían un subgrupo de mayor riesgo para desarrollar AVC.
No existen guías claras de manejo en estos pacientes. Según las series de casos publicadas ha sido universal el tratamiento con prednisona oral a todos los pacientes, iniciándose en dosis de 1mg/kg/día durante un mes para posteriormente iniciar dosis decreciente hasta 20mg/día y posteriormente ralentizar la bajada de la pauta hasta completar 1-2 años de tratamiento. Se han probado bolus de metilprednisolona, metotrexato, azatioprina o ciclofosfamida sin existir evidencia clínica a su favor. Se ha indicado también iniciar tratamiento antiagregante o anticoagulante concomitantemente. Se han descrito reinfartos cerebrales en las 6 primeras semanas tras el inicio de la terapia corticoidea y principalmente en el territorio vertebrobasilar, constituyendo la principal causa de muerte precoz en estos pacientes3,4,12. Esta infausta evolución precoz sería todavía más frecuente en los pacientes con afectación intracraneal a pesar de recibir tratamiento con prednisona e inmunosupresor2.
Se ha descrito la posible evolución hacia una demencia multiinfarto, a veces ya presente al iniciarse la enfermedad5,7,13,14. En algún caso se ha descrito cierta mejoría de la demencia tras el tratamiento corticoideo.
La estenosis de ambas arterias vertebrales es una situación muy poco reportada en la literatura médica y todavía más infrecuente asociando estenosis de carótidas. No se conoce la evolución de estas estenosis en la literatura especializada. Únicamente hemos encontrado un caso con determinación por angio-RMN de estenosis de carótida interna izquierda reperfundida 2 semanas después del inicio de la corticoterapia15.
