




La incidencia de la esclerosis sistémica (ES) en los ancianos es desconocida y según algunos estudios es del 1 por 1.000, sugiriendo los autores que esta enfermedad es probablemente infradiagnosticada en este grupo de población debido a un curso más favorable y a los menores cambios cutáneos que se atribuyen al envejecimiento o secundarios a otras patologías. Hay estudios que sugieren que la forma limitada de la ES es la más frecuente en los ancianos1,2.
La hipertensión arterial pulmonar (HAP) se desarrolla en más del 15% de los pacientes con ES. Esta complicación puede ocurrir de forma aislada en ausencia de enfermedad intersticial pulmonar y es más frecuente en la ES de tipo limitado asociándose con la presencia de anticuerpos anticentrómero en el 70–80% de los casos3.
Presentamos el caso de una mujer de 92 años con antecedentes de artrosis y artritis desde hace años que trataba con AINES y cambios de coloración en las manos que refería como cianosis ante la exposición al frío. Independiente para las actividades básicas de la vida diaria, sin deterioro cognitivo y deambulación con bastón limitada por artrosis y últimamente por disnea.
Remitida a urgencias por disnea progresiva de 1 mes de evolución, edemas en MMII y disminución de la diuresis.
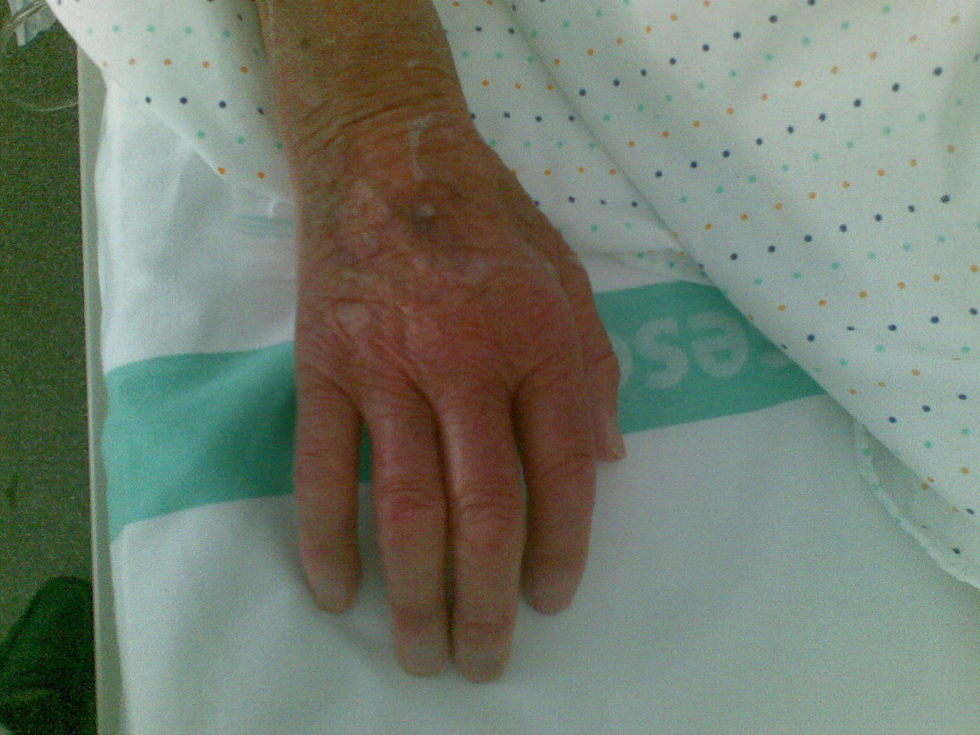
En la exploración física destacó disnea de reposo, trabajo respiratorio importante, saturación del 70–75% con oxigenoterapia a alto flujo y reservorio a 12l, bajo nivel de conciencia, hipotensión arterial, cianosis labial y en manos y esclerodactilia (fig. 1). No se evidenció ingurgitación yugular. En la auscultación cardiaca tonos rítmicos y un soplo sistólico panfocal. En la auscultación pulmonar hipofonesis bilateral y crepitantes. En MMII edemas hasta porción proximal de muslos.

La radiografía de tórax evidenció cardiomegalia y derrame pleural bilateral. El electrocardiograma mostró ritmo sinusal a 75lpm, HARI y T negativas en derivaciones inferiores y precordiales izquierdas. En la analítica destacó una hemoglobina de 11,6, tiempo de protrombina del 50%, dimero D de 0,5, urea de 101, creatinina de 1,39, potasio de 5,6, troponina de 0,11 y CKMB de 2,9. La TC torácica descartó signos de tromboembolismo pulmonar.
El ecocardiograma transtorácico mostró un ventrículo izquierdo de tamaño normal con hipertrofia concéntrica leve sin alteraciones de la contractilidad y función sistólica normal destacando una insuficiencia tricúspide moderada-severa con una presión sistólica en la arteria pulmonar (PSAP) de 61mmHg, HAP grave y cavidades derechas ligeramente dilatadas.
Por probable fenómeno de Raynaud y con la sospecha de HAP asociada a alguna enfermedad reumática autoinmune, se solicitaron anticuerpos antinucleares observándose un patrón centromérico con titulo 1/640.
La TC torácica de alta resolución objetivó cardiomegalia y signos de hipertensión pulmonar sin fibrosis pulmonar.
Ante el diagnóstico de HAP probablemente asociada a ES limitada se inició tratamiento con vasodilatadores (IECAS y amlodipino) y posteriormente con bosentan con una dosis inicial de 62,5mg cada 12h sin mejoría clínica, falleciendo la paciente a los 20 días de ingreso.
La HAP puede ser asintomática en las etapas tempranas y la aparición de síntomas indica enfermedad avanzada, asociada con un pobre pronóstico. La mortalidad por HAP asociada a la ES supera el 50% a los 12 meses3.Los síntomas más frecuentes son disnea primero con el ejercicio y después incluso en reposo. El fenómeno de Raynaud es el síntoma inicial casi en el 100% de los pacientes1. Las pruebas de función respiratoria muestran una disminución aislada de la difusión de CO. La TC torácica de alta resolución muestra el parénquima pulmonar normal, con dilatación de la arteria pulmonar4.
El patrón de referencia para el diagnóstico de HAP es el cateterismo cardiaco. La ecocardiografía doppler es la técnica más utilizada para detectar precozmente la HAP en pacientes con ES. La OMS recomienda la realización anual de ecocardiograma en los pacientes con esclerodermia5. Cuando la velocidad de regurgitación tricuspídea es mayor de 3m/s existe alto riesgo de hipertensión pulmonar. La normalidad de las pruebas de función respiratoria indican bajo riesgo de progresión a enfermedad pulmonar severa, mientras una disminución aislada en la capacidad de difusión de CO identificaría a la población de alto riesgo. Una DLCO inicial menor del 50% puede predecir el riesgo de desarrollo de HAP en más de 5 años6.
Las medidas terapeúticas incluyen oxigenoterapia, anticoagulación , diuréticos, vasodilatadores, prostaglandinas y el transplante pulmonar. El epoprostenol y el iloprost han demostrado su eficacia. El bosentan, un antagonista de los receptores de la endotelina, en dosis de 125mg/12h es un tratamiento eficaz de la HAP en ES; este efecto benéfico se puede mantener hasta 1 año3–5.
