




Este trabajo fue financiado por proyectos DIUC (Universidad de Concepción) y Fundación Andes otorgados al autor.
INTRODUCCIÓN
La enfermedad de Alzheimer (EA) es una enfermedad neurodegenerativa, caracterizada por la pérdida progresiva e irreversible de la memoria, la orientación y el razonamiento, lo que finalmente termina con las funciones intelectuales y la vida del individuo1.
La EA se caracteriza patológicamente por una degeneración neuronal selectiva que se manifiesta como una pérdida de las conexiones sinápticas2, además de la presencia de agregados proteicos (ovillos neurofibrilares [ON] y placas seniles [PS]) en la corteza cerebral y el hipocampo1. El comienzo y el desarrollo de la EA estarían determinados por factores genéticos y/o factores ambientales.
Durante los últimos 20 años, los estudios enfocados a entender el desarrollo y la progresión de la EA han caracterizado tanto los aspectos morfológicos y bioquímicos de los constituyentes de las PS como los aspectos de la biología celular de algunos de sus componentes. A través de estos estudios, se ha establecido que las PS corresponden a estructuras heterogéneas formadas principalmente por el péptido β-amiloide (βA), que tiene 40-42 aminoácidos de extensión1. Por una parte, el estudio molecular de la EA ha generado información importante acerca de la biología del péptido βA, y por otro lado numerosa información sobre las posibles causas de esta enfermedad. De esta forma, y utilizando como base la evidencia obtenida en estudios genéticos, bioquímicos y de biología molecular, se ha originado la hipótesis de «la cascada del amiloide», que propone a la generación y la formación de oligómeros neurotóxicos del péptido βA como los elementos principales en el desarrollo de la EA3.
EL PROCESAMIENTO PROTEOLÍTICO DE LA PROTEÍNA PRECURSORA DEL AMILOIDE (PPA) Y EL ORIGEN DEL PÉPTIDO β-AMILOIDE
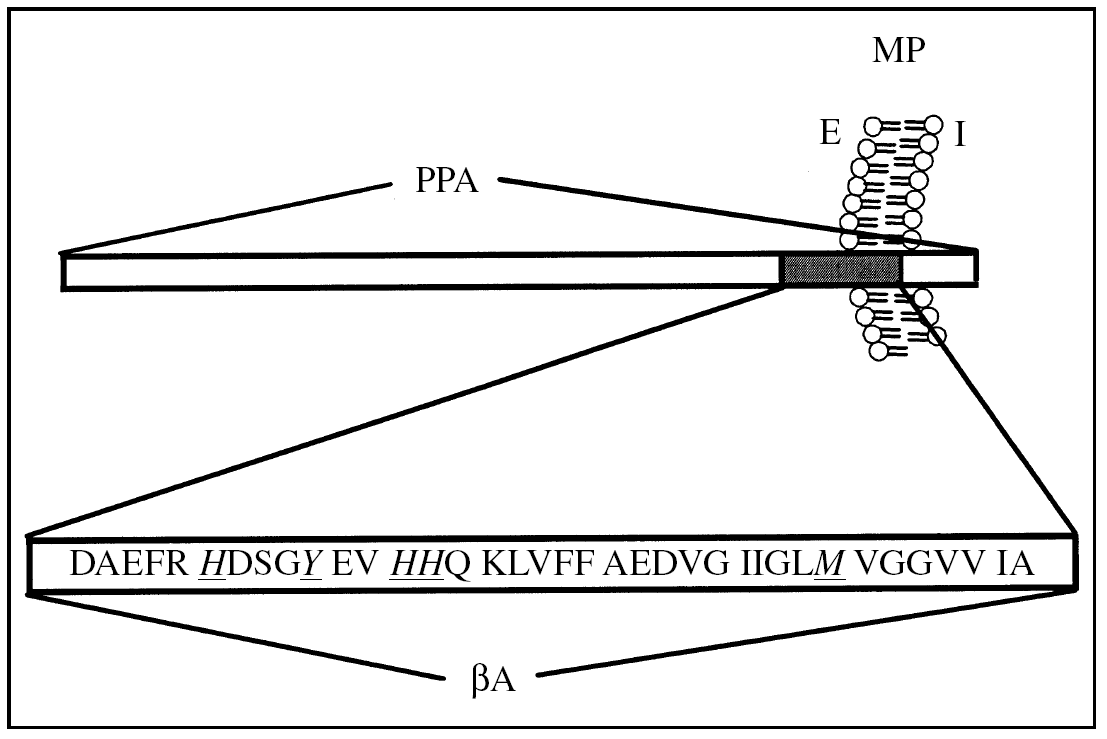
La PPA es una glucoproteína tipo I de transmembrana4,5 (fig. 1), que se expresa como distintas isoformas, lo que es regulado por un mecanismo de procesamiento alternativo6. Las isoformas del PPA 751 y 770, que contienen un dominio inhibidor de proteasas tipo Kunitz7, se expresan en forma ubicua, mientras que el PPA 695, que carece del dominio Kunitz, se expresa específicamente en el sistema nervioso central (SNC)8.
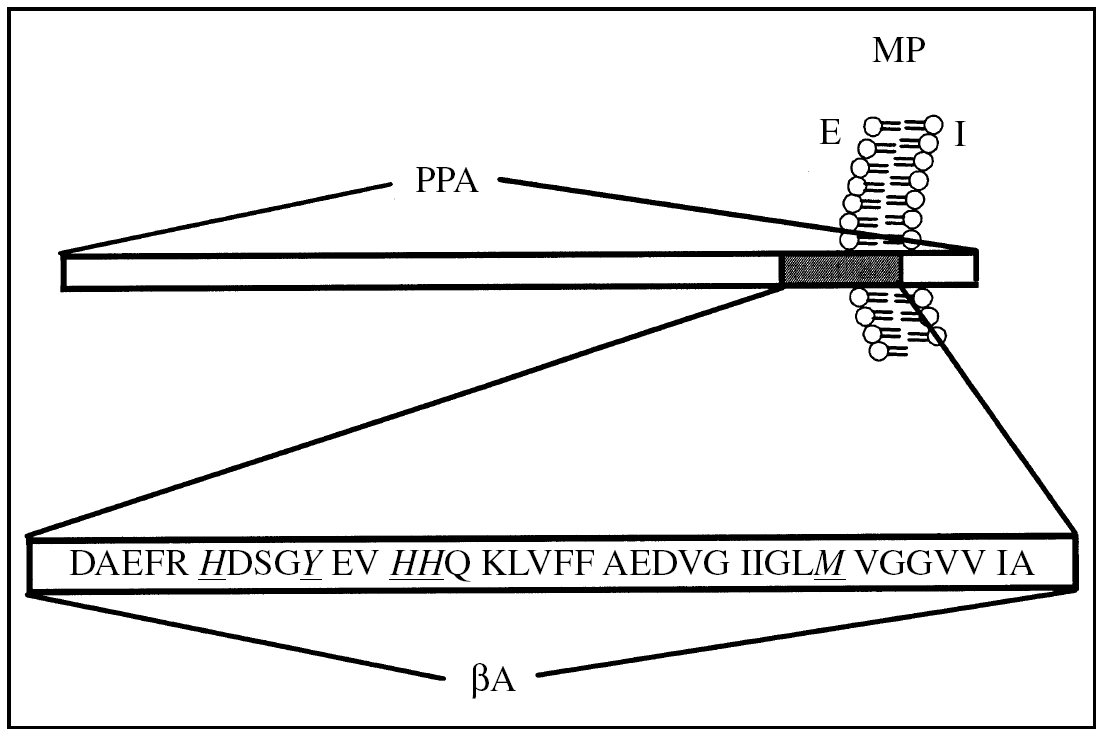
Figura 1.Esquema topológico de la proteína precursora de amiloide (PPA) y el péptido β-amiloide (βA). PPA es una proteína de transmembrana de tipo I. En gris se representa el péptido βA, que se genera a partir del corte proteolítico de la PPA por enzimas específicas (véase texto). En la secuencia aminoacídica del péptido βA, los aminoácidos destacados (H, Y y M) corresponden a aminoácidos involucrados en la coordinación y reducción del ion cobre. MP: membrana plasmática; E: medio extracelular; I: medio intracelular.
La PPA es procesada por proteasas denominadas secretasas9. La secretasa α corta la PPA en forma constitutiva dentro del dominio extracelular del péptido βA, generándose una PPA soluble no amiloidogénica (sPPA) y un fragmento truncado del βA. Alternativamente, la PPA puede ser cortada por la secretasa β en el dominio extracelular y por la secretasa γ en el dominio de transmembrana, con la consecutiva generación del péptido βA de 39 a 42 mer9.
Si bien la función fisiológica de la PPA neuronal o la de sus fragmentos es desconocida, estudios in vitro sugieren que la PPA asociada a la membrana y la PPA secretada tendrían un papel neuritogénico, en especial en: a) la adhesión celular; b) la extensión y formación de neuritas, y c) el transporte de vesículas sinápticas10-13. Además la PPA podría tener una función neurotrófica protegiendo a las neuronas frente a la lesión inducida por hipoglucemia, glutamato y daño oxidativo14-17.
POSIBLE PAPEL DE LA PPA EN EL TRANSPORTE DEL ION COBRE
El ion cobre es un micronutriente esencial para la fisiología humana, que se encuentra coordinado a la estructura de diversas proteínas, como las superóxido dismutasa (SOD), la citocromo C oxidasa y la ceruloplasmina. La homeostasis del ion cobre está altamente regulada por proteínas localizadas en diferentes compartimientos celulares como membrana plasmática, organelos y citoplasma18. En este contexto, es interesante que la PPA, que está localizada en la membrana plasmática y tiene sus sitios de unión a cobre orientados hacia el extracelular, pudiera participar en las etapas iniciales de la incorporación del metal desde el espacio extracelular al espacio intracelular19-22. La PPA presenta al menos dos sitios putativos de unión a cobre: uno localizado entre los aminoácidos 135 y 156 (PPA135-156)20-23, y el segundo localizado dentro de la secuencia que corresponde al péptido βA, dominio (PPA672-711)24,25 (fig. 1).
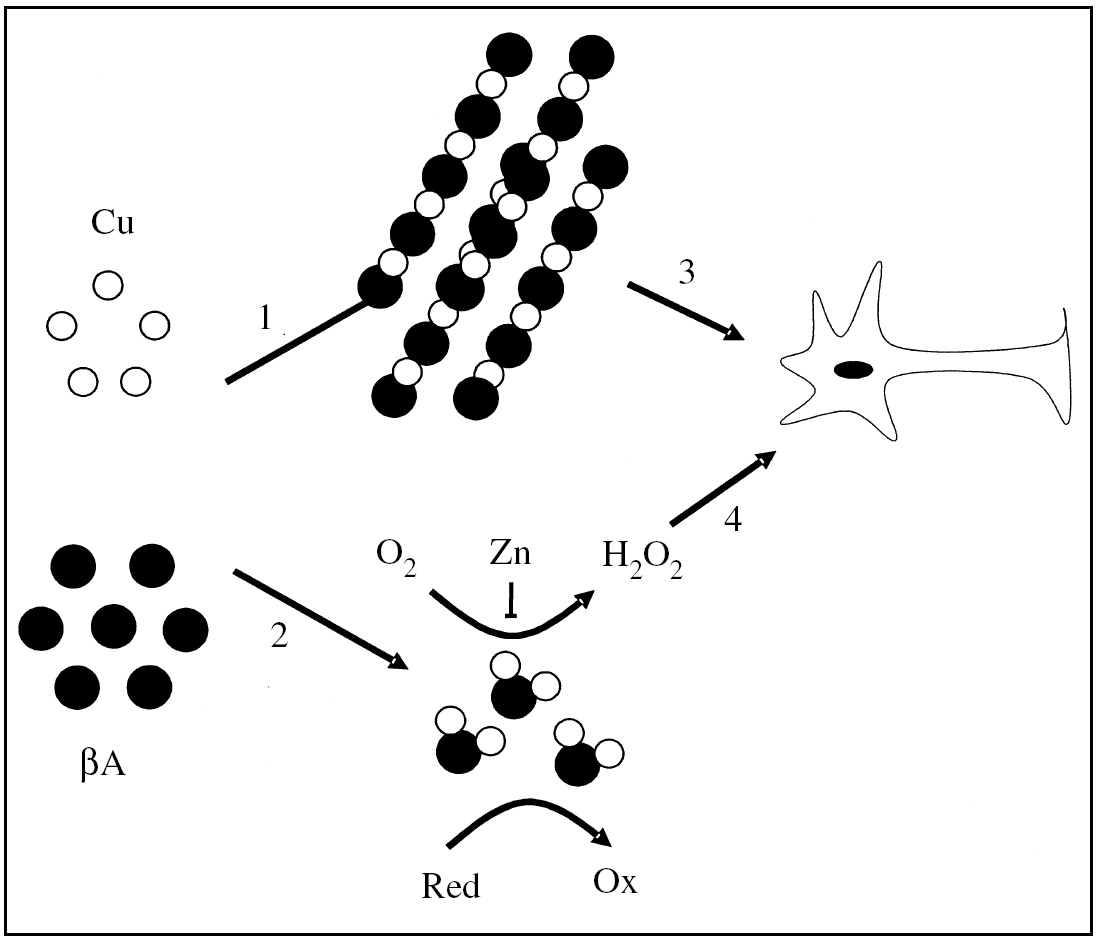
Considerando que el gen de la PPA tiene todas las características de un gen de expresión constitutiva y que la PPA es expresado en todos los tejidos de los mamíferos5, la capacidad reductora de cobre que la PPA presenta en la superficie de la membrana plasmática afectaría tanto a las células neuronales como a las no neuronales, favoreciendo la formación de complejos metal-proteína en el espacio intracelular. Por lo tanto, en células normales la actividad reductora de cobre de la PPA podría ser favorable, presentando el Cu(I) al transportador de Cu(I). En condiciones desfavorables, un aumento en los niveles de la PPA sobre la superficie celular incrementaría los niveles de cobre(I) y favorecería la formación de especies reactivas de oxígeno (ERO)25-27. En estas circunstancias, la PPA o el péptido βA, al coordinar el metal, adquirirían una actividad pro-oxidante26,28-30 (fig. 2), lo que podría explicar algunos aspectos del daño oxidativo que se presenta en la EA28,31. En un contexto celular, los efectos generados por la interacción de la PPA (PPA135-156) o el βA con cobre dependerían de la expresión y la localización subcelular de la PPA, así como de la generación del βA.
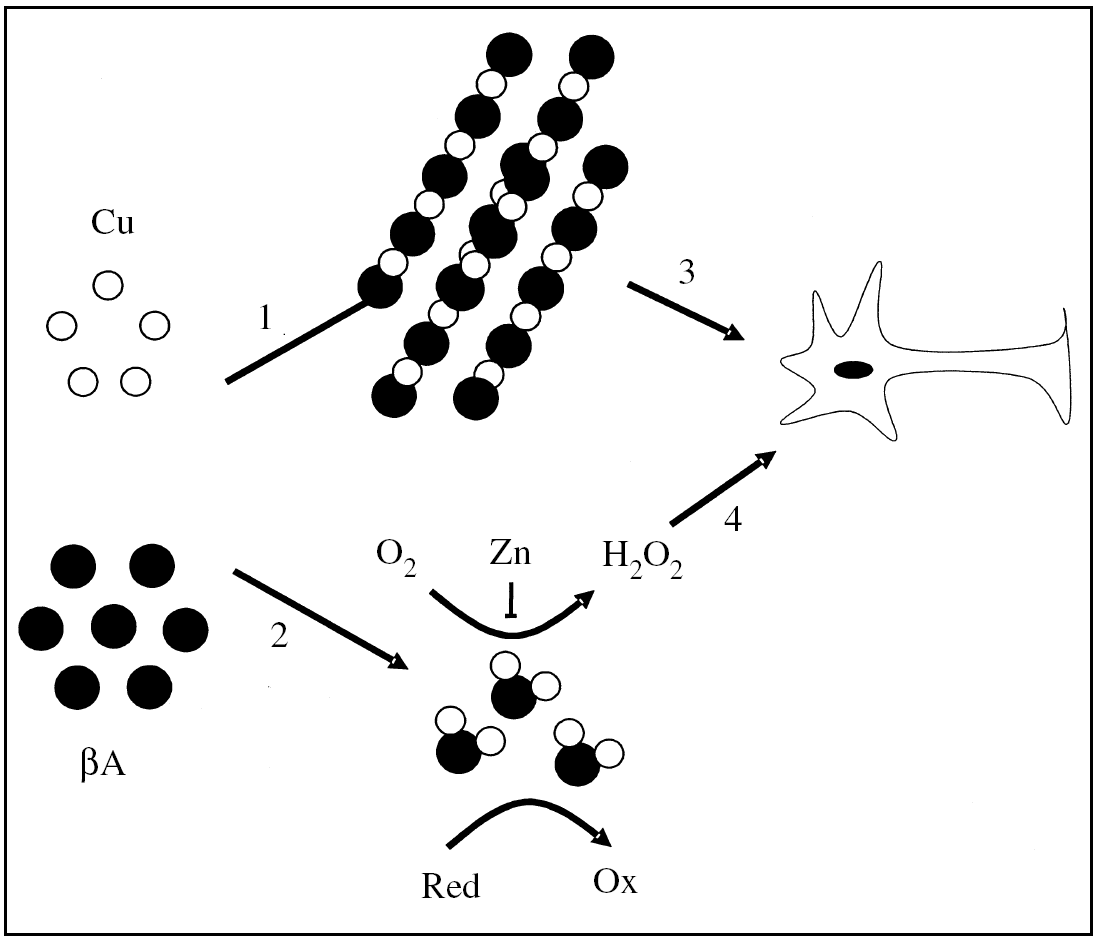
Figura 2.Modelo del posible papel del ion cobre sobre la neurotoxicidad del péptido β-amiloide en la enfermedad de Alzheimer. El ion cobre podría inducir la agregación del péptido βA (1), o inducir la formación de complejos βA-Cu solubles con propiedades redox (2). Los agregados podrían tener una acción tóxica directa sobre las neuronas (3). De forma alternativa, los complejos βA-Cu podrían generar peróxido de hidrógeno, que podría desencadenar daño oxidativo en la membrana plasmática o en el espacio intracelular de la neurona (4). Esta última reacción podría ser inhibida parcialmente por el ion cinc. Red: agente reductor; Ox: agente oxidado.
EL PÉPTIDO βA COMO ELEMENTO CENTRAL EN LA PATOLOGÍA DE LA EA
El péptido βA se acumula en la corteza cerebral de pacientes que presentan EA, ya sea en forma de PS en el parénquima cerebral o en forma de depósitos congofílicos en la vasculatura cerebral. El péptido βA1-40 es la especie soluble que se encuentra en mayor abundancia en fluidos biológicos como el plasma y el líquido cefalorraquídeo32, mientras que el péptido βA1-42, que es la especie del βA menos abundante, se acumula de forma particular en las PS presentes en el cerebro de pacientes con EA33,34.
Todavía no se conocen todos los factores involucrados en el comienzo y el desarrollo de la EA. Al respecto, se han identificado 3 genes asociados al desarrollo de la EA, que al estar mutados favorecerían el inicio temprano de esta enfermedad. Las mutaciones en estos genes (PPA, presenilina-1 y presenilina-2) conducen a la aparición temprana de depósitos de amiloide, aumento en la concentración total del βA e incremento relativo de la concentración del βA1-42 respecto a las otras especies de menor peso molecular como el βA1-4035,36. Sin embargo, las mutaciones en estas proteínas sólo explicarían un bajo porcentaje de la enfermedad (~10%)1, lo que sugiere que otros factores genéticos y no genéticos determinarían la aparición de esta enfermedad.
El βA es conocido por ser neurotóxico en el rango micromolar37. Sin embargo, aún no ha sido dilucidado el mecanismo por el que el βA es neurotóxico, aunque es probable que sea mediado en parte por la formación de especies reactivas de oxígeno (ERO)38 (fig. 2). Recientemente se ha descrito que el péptido βA une cobre con una alta afinidad24 y presenta en su estructura un sitio de coordinación cooperativa y alostérico por este metal39. El complejo βA-Cu tiene un potencial de óxido-reducción positivo y produce H2O2 desde el O2 a través de la reducción de Cu (II) a Cu (I)25,26,29,30. En esta reacción, aminoácidos como las histidina 13 y 14 tendrían un cierto papel en la coordinación del metal, y aminoácidos como la tirosina 10 y la metionina 35 ejercerían un posible papel en la transferencia de electrones desde el péptido βA al ion cobre39 (fig. 1). El H2O2 que es generado directamente por el complejo βA-Cu podría contribuir a la neurotoxicidad que el βA genera sobre cultivos primarios26,30 y a la oxidación de moléculas biológicas que se detecta en la corteza de pacientes con EA26,30,31 (fig. 2). Este patrón de daño oxidativo también se ha observado en ratones transgénicos (PPA 2576) que sobreexpresan el PPA, donde es posible detectar PS enriquecidas en hierro y en moléculas oxidadas40,41.
Considerando que los niveles de cobre, hierro y cinc se encuentran elevados en las PS de pacientes con EA42,43, y que estos metales potencian la neurotoxicidad del βA26,30, se ha sugerido que quelantes hidrofóbicos de metales podrían servir como agentes terapéuticos para tratar a los pacientes con EA. Al respecto, hay evidencia experimental que apoya este planteamiento. El clioquinol (CQ), quelante del cobre y cinc, inhibe la formación de los depósitos de βA que se presentan en el cerebro de ratones transgénicos que sobreexpresan la PPA44. De la misma forma, en estudios preliminares realizados en humanos, se ha observado que la administración de esta sustancia a pacientes con EA disminuye el deterioro cognitivo45.
METALES DE TRANSICIÓN, AGREGACIÓN Y NEUROTOXICIDAD DEL PÉPTIDO βA
Los agregados amiloides formados a partir del péptido βA presentan propiedades fisicoquímicas características como: a) birrefringencia a la luz polarizada después de ser teñidos con rojo Congo; b) patrón de difracción de rayos X característico, y c) aspecto fibrilar en imágenes registradas por microscopia electrónica46,47.
La formación de los agregados de amiloide depende de variables que afectan el tipo de estructura secundaria que el péptido βA adopta en solución, como pH, temperatura, solvente y concentración peptídica48-50. Los dominios hidrofóbicos (17-21; 39-42) y los dominios hidrofílicos (12-15) del βA serían de gran importancia para la formación de los agregados de amiloide51-53.
Los agregados del péptido βA tienen un efecto neurotóxico en el rango de concentración micromolar37. Estos efectos tóxicos podrían estar mediados por una acción directa sobre la membrana celular54-58 (fig. 2). De hecho, el péptido βA es capaz de interaccionar con elementos que componen la membrana plasmática como receptores de membrana55,58, colesterol, fosfolípidos y gangliósidos57,59. Esta interacción directa de la βA con la membrana puede incluso resultar en la formación de poros que permiten el paso de iones calcio al interior de la célula56.
Sumado a esto, se ha descrito que el βA presenta una estructura química que facilita la formación de ERO60, lo que podría inducir la oxidación de proteínas y lípidos localizados en la membrana plasmática alterando la función celular61,62. En estos procesos intervendrían metales como hierro y cobre, que podrían catalizar la generación de especies reactivas de oxígeno vía las reacciones de Fentom o Haber-Weiss25,29.
Además las ERO generadas por la acción del péptido βA facilitarían su agregación, lo que se potenciaría en presencia de metales como hierro y cobre. Por lo tanto los metales tendrían un efecto dual: a) promoviendo la agregación del péptido βA, y b) potenciando su acción tóxica vía la generación de ERO63 (fig. 2). Hay evidencia experimental que apoya la participación de metales como el ion cobre en el mecanismo de toxicidad de los agregados del péptido βA. Al igual que el hierro, este metal se encuentra acumulado en las PS43, y tiene la capacidad de inducir la agregación del péptido βA in vitro e in vivo64,65. En conclusión, el ion cobre podría participar en el mecanismo de daño oxidativo, catalizando la formación de ERO, promoviendo la agregación del péptido βA y potenciando los efectos neurotóxicos de los agregados de βA (fig. 2).
Estudios previos han sugerido que el péptido βA formaría complejos solubles al coordinar cobre o cinc66,67, y esto podría ser modulado por la concentración de los metales, así como por factores tales como pH y fuerza iónica24,64. La estequiometría y la identidad del metal asociado al péptido βA serían determinantes en las propiedades redox de los complejos bioinorgánicos resultantes28. Además, la toxicidad generada por los complejos solubles e insolubles dependería del sistema antioxidante que posea la célula blanco.
De esta forma, los complejos bioinorgánicos formados por la asociación de los metales de transición que se acumulan en las placas seniles63,69 y el péptido βA generarían el daño oxidativo que se detecta en el cerebro de pacientes con EA31.
La idea de que los metales de transición tendrían un papel en la EA se ha visto reforzada por estudios recientes que muestran que los niveles del ion cobre se encuentran aumentados en el plasma de pacientes con EA70-72, sugiriendo que los pacientes con esta enfermedad presentan alteraciones en la homeostasis de este ion metálico. Además, estudios preliminares muestran que la agregación del péptido βA podría ser acelerada por metales de transición vía un mecanismo oxidativo catalizado por metales como el hierro63. Además, hemos encontrado que las fibras de amiloide son capaces también de reducir el cobre, lo que sugiere que las ERO podrían ser generadas durante etapas iniciales o en etapas tardías del proceso de agregación25.
En el contexto de la EA se puede formular la hipótesis de que la formación de H2O2 por el complejo βA podría sobrepasar las defensas celulares antioxidantes (catalasa, glutatión peroxidasa). El H2O2 podría difundirse a través de las membranas biológicas, y de esta forma ser la fuente de daño oxidativo que se detecta en diversos compartimientos celulares afectados en el cerebro de pacientes con la EA40,73-76. Por lo tanto, el daño oxidativo presente en la EA podría ser consecuencia de la sobreproducción de H2O2 por fuentes biológicas como el complejo βA-Cu. Además, la actividad oxidasa del βA-Cu podría generar un daño secundario como resultado de la oxidación y consumo de sustratos biológicos importantes para diversos procesos biológicos como la vitamina C, las catecolaminas y el colesterol.
No se han determinado los factores que favorecen la formación del complejo βA-Cu. Sin embargo, se podría especular que un incremento en los niveles de cobre en la corteza cerebral aumentaría la probabilidad de formación de este tipo de complejos neurotóxicos. De hecho, se ha establecido experimentalmente que hay una correlación directa entre los niveles cerebrales del ion cobre y la edad de los ratones de experimentación, lo que es modulado por la expresión de la PPA y el péptido βA77-79. Esto indica que el envejecimiento podría favorecer la formación de este tipo de complejos y por lo tanto el desarrollo de esta enfermedad. Es interesante destacar que existe una mayor susceptibilidad a desarrollar la EA al envejecer. Además se ha encontrado que hay un incremento de la concentración de agentes reductores en la EA80. Este incremento en equivalentes reductores podría corresponder a una compensación en respuesta al incremento del daño oxidativo, como la regulación positiva de la enzima glucosa-6-fosfato deshidrogenasa, que regula los niveles de glutatión intracelular y cuya actividad es aumentada en la EA80. En estas condiciones, un incremento inapropiado de equivalentes reductores se acompañaría de un incremento en los valores de H2O2, lo que generaría un efecto paradójico al inducir un mayor aumento en la formación de H2O2 catalizado por el complejo βA-Cu, y de esta forma originar un círculo bioquímico vicioso.
El papel del ion cinc en la patología de la EA es, al parecer, complejo. El ion cinc induce la precipitación del βA para formar parte de las placas de amiloide42,81,82, y ya que el ion cinc es capaz de suprimir parcialmente la formación de H2O2 formado por el βA83, se ha sugerido que las placas seniles podrían corresponder a un sistema de defensa celular, donde el ion cinc precipita el péptido βA inhibiendo su actividad oxidasa83 (fig. 2). Esto podría explicar la existencia de una correlación inversa entre el tamaño de las PS y el daño oxidativo mediado por H2O2, presente en el cerebro de pacientes con EA83. Sin embargo, se ha encontrado que agregados del péptido βA purificado desde PS coordinan una cantidad de ion cinc insuficiente para inhibir su actividad oxidasa30. De hecho, a pesar de la correlación inversa entre el daño oxidativo y el tamaño de la PS, son justamente las PS de mayor tamaño las que presentan mayor concentración de productos oxidados83. La precipitación del péptido βA por el ion cinc podría también inhibir la eliminación y el catabolismo del βA en el cerebro82 y de esta forma favorecer la neurotoxicidad de los agregados del βA.
Por lo tanto, y a la luz de los elementos expuestos en esta revisión, la interacción del péptido βA con los metales de transición podrían ser parte del mecanismo responsable del deterioro cognitivo que presentan los enfermos con EA44,45.
