




Paciente de 88 años que ingresó en el servicio de medicina interna por cuadro febril de 48h de evolución acompañado de postración y dolor generalizado de predominio a nivel de cintura escapular, codos y manos con impotencia funcional marcada. La paciente tenía como antecedentes más importantes hipertensión arterial, diabetes mellitus tipo 2, obesidad, insuficiencia renal crónica de grado leve, claudicación intermitente y depresión. Se encontraba en seguimiento por ginecología por episodios de metrorragias con relación al engrosamiento endometrial, con legrado en 2 ocasiones cuya anatomía patológica no había resultado concluyente para carcinoma endometrial. En cuanto a su situación basal, la paciente no presentaba deterioro cognitivo, y desde el punto de vista funcional, caminaba con andador, siendo parcialmente dependiente para las actividades de la vida diaria.
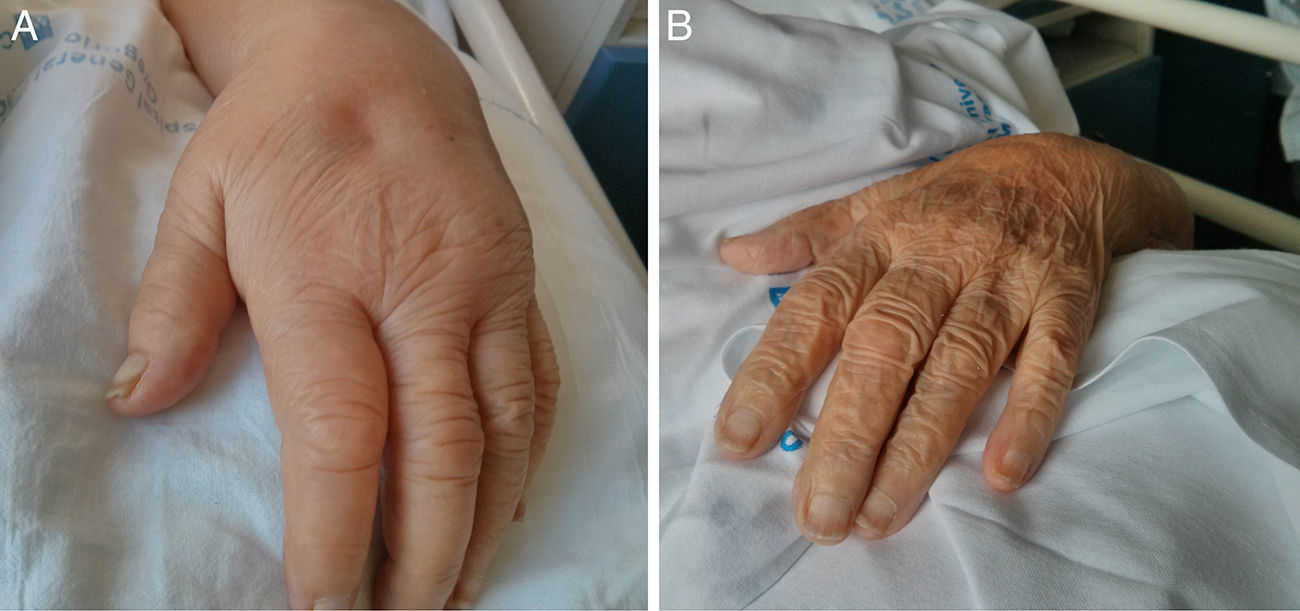
En la exploración destacaba una marcada debilidad en ambas cinturas de predominio escapular, con imposibilidad para levantar activamente los brazos y dificultad intensa para la flexo-extensión de codos, muñecas y dedos. La movilidad pasiva también se encontraba limitada. Además, presentaba importante edema con fóvea a la presión digital localizado en el dorso de manos y dedos, bilateral y simétrico, sin eritema ni claro aumento de temperatura local (fig. 1A). La palpación de las articulaciones metacarpofalángicas e interfalángicas era dolorosa, no así la palpación de grupos musculares.
 Se observa importante edema con fóvea a nivel de la mano izquierda. B) Evolución tras 48h de tratamiento: desaparición del edema de la mano.")
En las pruebas complementarias destacaba la presencia de anemia leve normocítica-normocrómica (con cifra de hemoglobina de 10,6g/dl), velocidad de sedimentación globular (VSG) de 120mm 1.ª hora, proteína C reactiva (PCR) 15mg/dl (valor de referencia <0,5mg/dl). Las enzimas musculares: AST, CK, LDH y aldolasa, el perfil tiroideo y la uricemia fueron normales. Con respecto al perfil de autoinmunidad, tanto el factor reumatoide (FR) como el anticuerpo antipéptido cíclico citrulinado (anti-CCP), los anticuerpos antinucleares (ANA), los anticuerpos anticitoplasma de neutrófilo (ANCA) y las crioglobulinas resultaron negativas, y los niveles de complemento fueron normales. Los biomarcadores (alfa-fetoproteína, antígenos CA-125, CA-15-3, CA-19-9 y CYFRA 21-1) fueron negativos. La radiografía de tórax y la ecografía abdominal no mostraron hallazgos patológicos reseñables.
El diagnóstico diferencial inicial incluyó fundamentalmente la polimialgia reumática, la artritis reumatoide del anciano y el síndrome de sinovitis simétrica seronegativa remitente con edema y fóvea (remitting seronegative symmetrical synovitis with pitting edema o síndrome RS3PE). Se descartó la posibilidad de artritis reumatoide con relación a la negatividad para FR y anticuerpos anti-CCP. El edema de manos característico y la presencia de afectación sinovial apoyaban el diagnóstico de síndrome RS3PE sobre la polimialgia reumática.
Se inició tratamiento esteroideo con prednisona 15mg al día, observándose una respuesta espectacular. Tras la primera dosis, la paciente era capaz de movilizar espontáneamente ambos miembros superiores, aunque aún con ángulo de movilidad reducido. Tras la segunda dosis de prednisona, la movilidad de ambas cinturas se recuperó casi por completo, y el edema de manos se resolvió (fig. 1B). Cinco días después de iniciado el tratamiento, la paciente pudo ser dada de alta a domicilio, con buena recuperación funcional que le permitía por ejemplo, comer sola y asearse la cara, e importante disminución del dolor poliarticular.
Descrito por primera vez en 1985 por McCarty et al. en una serie de 10 pacientes1, el síndrome RS3PE afecta preferentemente a varones ancianos. La sintomatología se inicia con el compromiso de la mano en forma de tumefacción dolorosa de muñecas, articulaciones metacarpofalángicas e interfalángicas que progresa de forma rápida, típicamente en menos de un mes, hacia el edema franco con fóvea en dorso de manos y falanges y con importante limitación funcional de las mismas y extensión también a nivel de cintura escapular. La afectación de tobillos y pies también ha sido descrita. El dolor sería la traducción clínica de la polisinovitis, y el edema con fóvea se produciría por aumento de la permeabilidad de los capilares de los tejidos blando y celular subcutáneo, consecuencia de la tenosinovitis de los músculos extensores de la mano2,3. Actualmente, el diagnóstico del RS3PE se apoya en 7 criterios propuestos por Olivo en 1994: edad igual o mayor a 65 años; factor reumatoide negativo; polisinovitis simétrica afectando a muñecas, metacarpofalángicas, interfalángicas proximales y vainas tendinosas de las manos; edema «en piel de naranja» con fóvea en las zonas afectadas; rigidez matutina; rápida respuesta al tratamiento esteroideo, y exclusión de otras enfermedades4.
Los hallazgos de laboratorio suelen mostrar alteraciones inespecíficas con aumento de reactantes de fase aguda. Es común el hallazgo de una VSG y una PCR altas, con FR y anticuerpos anti-CCP negativos. Los ANA son habitualmente negativos o positivos a títulos bajos y, a veces, pueden presentar una anemia normocítica normocrómica de trastornos crónicos2,5.
En cuanto a la etiopatogenia del síndrome, sigue siendo desconocida. Se han propuesto factores medioambientales, genéticos e incluso infecciosos que podrían tener un papel en el desarrollo del síndrome. Se encuentra asociación con el antígeno HLA-B7 en aproximadamente 50-60% de los pacientes, lo que podría plantear un factor genético en su patogénesis2,6,7. También se ha descrito la relación entre el inicio del síndrome y la exposición a agentes infecciosos como el parvovirus B19 y el bacilo de Calmette-Guérin7.
Aunque el síndrome RS3PE puede presentarse de forma aislada, existen descripciones en la literatura de casos asociados a diferentes procesos inflamatorios y neoplasias malignas4–6. Así, se ha descrito asociado a adenocarcinomas de colon, estómago, endometrio y próstata; carcinomas de mama, ovario, pulmón, vejiga, hepatocelular y de páncreas, y tumores hematológicos como linfoma de Hodgkin, leucemia linfática crónica y síndromes mielodisplásicos5,6,8–10. Existe evidencia de que los niveles incrementados de VEGF y de IL-6 en estos tumores, especialmente en los de estirpe glandular, tendrían un papel importante en la patogénesis del edema del síndrome RS3PE5,7,8. Cuando se manifiesta como síndrome paraneoplásico, tiende a presentar manifestaciones sistémicas tales como astenia, fiebre y pérdida de peso. Por otra parte, se han descrito casos aislados asociados a otras enfermedades reumáticas, como la sarcoidosis, poliarteritis nodosa, amiloidosis y espondilitis anquilosante, entre otras5.
La respuesta del RS3PE al tratamiento con corticoides a dosis baja es precoz. La dosis habitual oscila entre los 10-20mg/día inicialmente, para disminuir paulatinamente la dosis en un periodo de tiempo de entre 6 a 18 meses. Tras las primeras dosis, la mejoría clínica es evidente, con disminución del edema hasta su desaparición, y con recuperación de la movilidad y mejoría o incluso desaparición del dolor. Los antiinflamatorios no esteroideos o la cloroquina podrían estar indicados en determinados casos2,3,5.
El pronóstico cuando se presenta como síndrome aislado es bueno. La remisión completa suele alcanzarse en menos de un año y, aunque pueden producirse recidivas, estas no comprometen el pronóstico. En los casos que se manifiesta como síndrome paraneoplásico, la respuesta a corticoides puede ser más pobre, y el pronóstico se relaciona con la enfermedad tumoral2,4.
En conclusión, presentamos un caso de síndrome RS3PE en una paciente de edad avanzada, con alta sospecha previa de adenocarcinoma endometrial no confirmada histológicamente, que respondió favorablemente y de forma rápida a dosis bajas de prednisona.
