




La enfermedad de Erdheim-Chester es una proliferación anormal de histiocitos no-Langerhans, de etiología desconocida, que casi invariablemente aparece en edades adultas. Presenta una ligera predilección por el sexo masculino, y el compromiso óseo es característico. El 50% de los casos cursan con enfermedad sistémica que afecta frecuentemente el corazón, los pulmones, los riñones, el retroperitoneo, el sistema nervioso central y la piel. Se presentan dos pacientes de sexo masculino, con edades de 61 y 22 años, respectivamente, ambos con afección del sistema óseo. Histológicamente, en ambos casos se observó una proliferación de células fusiformes y de histiocitos espumosos. En la investigación inmunohistoquímica, las biopsias mostraron proliferación histiocítica negativa para CD1a y S100, y positiva para CD68.
Erdheim-Chester Disease is a rare systemic xanthogranulomatous disease of unknown aetiology in which there is an abnormal proliferation of mononuclear non-Langerhans cell histiocytes. It affects both sexes, with a slight male predominance, and usually occurs in middle age, between the fifth and sixth decade of life. Although long bone involvement is almost universal, 50% of patients also have extra skeletal manifestations, heart, lungs, kidneys, retroperitoneal space, central nervous system and skin being the most frequent involved sites. We present two cases of Erdheim-Chester disease in male patients aged 61 and 22 years, both of whom had bone lesions. A proliferation of spindle cells and foamy histiocytes was present in both cases. In all the biopsies, immunohistochemistry showed histiocytes positive for CD68 and negative for CD1a and S100.
La enfermedad de Erdheim-Chester es una forma rara de proliferación anormal de histiocitos no-Langerhans, de etiología desconocida1,2, que casi invariablemente se presenta en pacientes de edad adulta, con un pico a nivel de la quinta y la sexta décadas de la vida. Aunque es padecida por ambos sexos, tiene una ligera predilección por el sexo masculino. En el aspecto clínico se observan múltiples signos y síntomas, dentro de los cuales la afección ósea con predilección por huesos largos se observa en todos los casos3. El 50% de los casos presentan enfermedad sistémica, y los sitios de compromiso más frecuentes son el corazón, los pulmones, los riñones, el espacio retroperitoneal, el sistema nervioso central y la piel. El pronóstico depende de la agresividad del cuadro clínico2.
En la presente revisión mostramos 2 casos con cursos clínicos diferentes. Uno de ellos es semejante a lo descrito en la literatura, mientras que en el otro, de reciente diagnóstico, el cuadro clínico es menos característico.
Presentación de casosCaso 1Paciente masculino de 61 años de edad que a los 33 años presentó fractura de la pierna derecha y posteriormente, a los 45 años, se fracturó la izquierda. Además, con el correr de los años a su cuadro se agregó diabetes insípida, hipertensión arterial, prolapso e insuficiencia mitral, esclerosis ósea a nivel de la tibia, y reducción de los cartílagos de las articulaciones de los miembros inferiores.
Posteriormente, en el año 2009, presentó dolor crónico en ambas extremidades, que fue diagnosticado como artrosis. En julio del mismo año fue ingresado nuevamente por presentar febrícula vespertina (38°C), astenia de 2 meses de evolución y sudoración nocturna. En la TAC abdominopélvica se apreció a nivel de ambas uniones pieloureterales una lesión sólida hipodensa (de 3×2 y 2,5×1cm) derecha e izquierda, respectivamente, con dilatación de cálices renales, extendiéndose de forma difusa en la pared de la aorta abdominal a nivel subrenal. El cuadro clínico fue compatible con fibrosis retroperitoneal multifocal. El resto era normal.
Se realizó una laparoscopia exploratoria con biopsia retroperitoneal, en la cual se evidenció tejido adiposo maduro con infiltrado inflamatorio focal de linfocitos, plasmocitos e histiocitos, relacionándose con el cuadro clínico de fibrosis retroperitoneal multifocal.
En febrero de 2010 se inició la terapia con colchicina para la fibrosis retroperitoneal, manteniendo aún aumento del fibrinógeno y del dolor lumbar. En septiembre de 2010 se suspendió la terapia con colchicina. En noviembre presentó un episodio de diplopía secundaria a déficit del músculo recto inferior derecho.
En los estudios de imágenes se observó engrosamiento del septo interventricular a nivel cardíaco y de la pleura a nivel del lóbulo inferior derecho. En la tomografía con emisión de positrones se observó, además, compromiso del maxilar derecho.
La gammagrafía ósea evidenció múltiples áreas hipercaptantes fuertemente sugestivas de enfermedad de Erdheim-Chester, con compromiso neurológico central, retroperitoneal, muy probablemente pulmonar y cardíaco.
Se realizó revisión de la biopsia retroperitoneal, en la que se encontraron histiocitos negativos para S100 y CD1a y positivos para CD68; por lo tanto, se concluyó como histiocitosis de células no-Langerhans (enfermedad de Erdheim-Chester).
Se inició la terapia con interferón alfa, que se suspendió en febrero del año en curso por falta de respuesta terapéutica, y se instauró imatinib 100mg, una cápsula/día, pauta que se suspendió al cabo de 20 días.
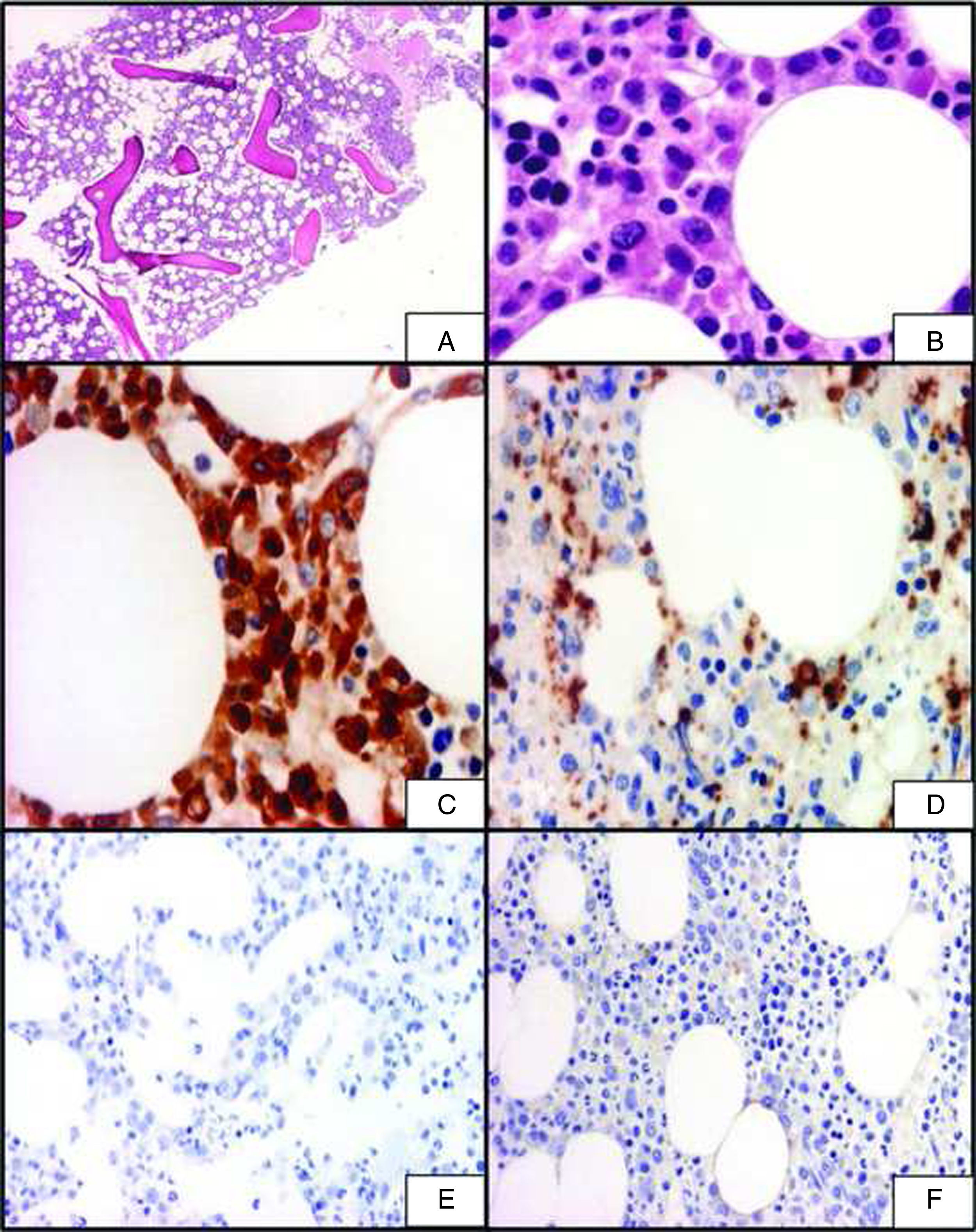
Además se realizó biopsia de médula ósea para valorar el diagnóstico, en la cual se observó leve hipercelularidad (fig. 1A) con representación tricelular normal (fig. 1B). Se apreció una proliferación intersticial de elementos histiocitarios CD68 y PGM1 positivos al estudio de la inmunohistoquímica y negativos para CD1a y S100 (fig. 1C-F), con focos de linfocitos bien diferenciados, inmunorreactivos para CD20 y CD3. Por lo tanto, se confirmó el diagnóstico de enfermedad de Erdheim-Chester.
 Sección de médula ósea en la que se observa una leve hipercelularidad (H&E, 4×). B) Ampliación de la microfotografía anterior en la que se aprecia una representación lineal normal con presencia de células histiocitarias (H&E, 40×). C) Inmunotinción para CD68 en la que se observan positivos los elementos histiocitarios (40×). D) Inmunotinción para PGM1: positividad en los histiocitos (40×). E) Inmunotinción para CD1a: negatividad en los elementos histiocitarios no-Langerhans (40×). F) Inmunotinción para S100 en la que se observa negativa la reacción en los elementos histiocitarios (40×).")
A) Sección de médula ósea en la que se observa una leve hipercelularidad (H&E, 4×). B) Ampliación de la microfotografía anterior en la que se aprecia una representación lineal normal con presencia de células histiocitarias (H&E, 40×). C) Inmunotinción para CD68 en la que se observan positivos los elementos histiocitarios (40×). D) Inmunotinción para PGM1: positividad en los histiocitos (40×). E) Inmunotinción para CD1a: negatividad en los elementos histiocitarios no-Langerhans (40×). F) Inmunotinción para S100 en la que se observa negativa la reacción en los elementos histiocitarios (40×).
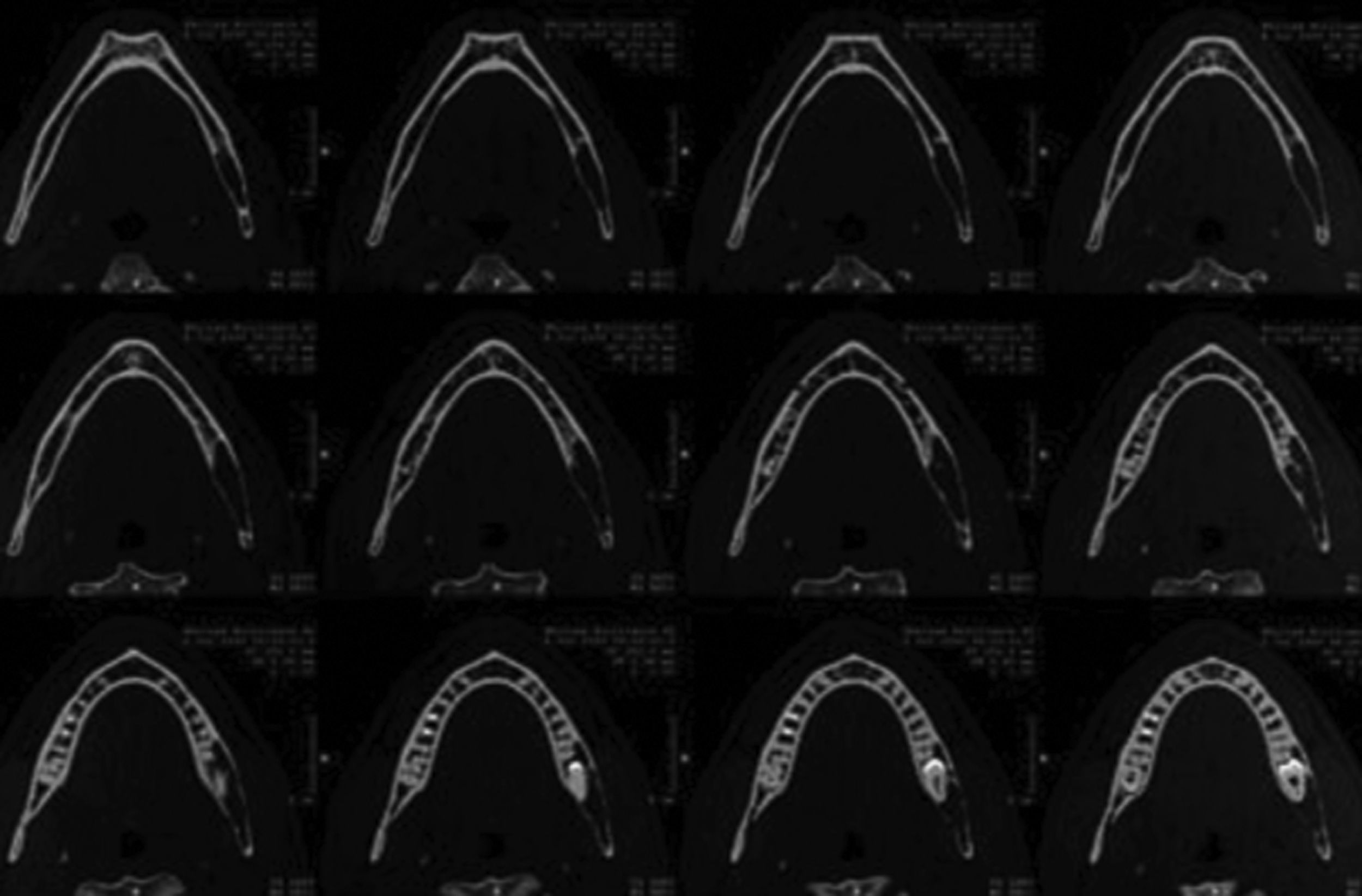
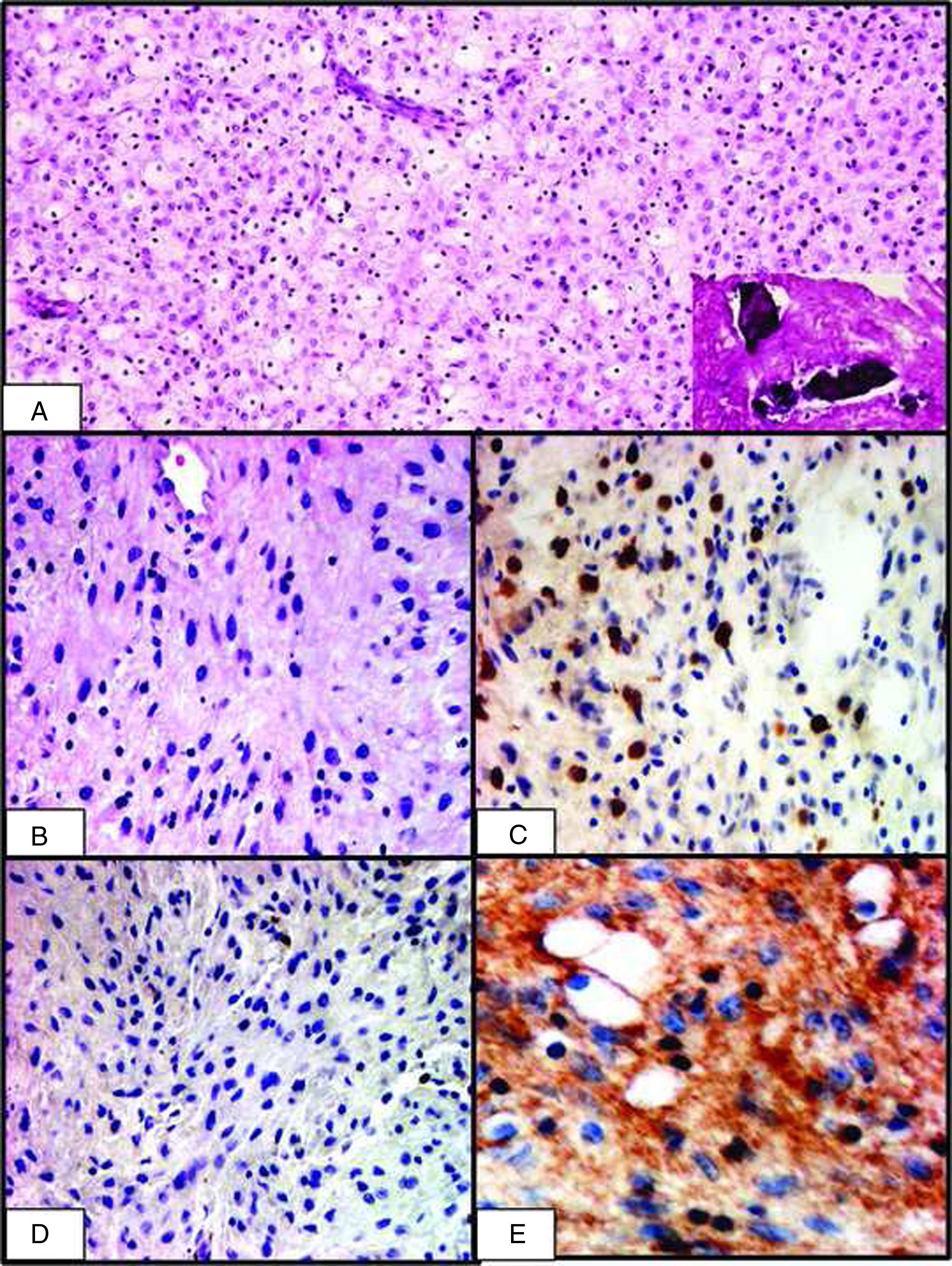
Paciente masculino de 22 años de edad que inició su cuadro clínico en junio del 2009, presentando parestesia del hemilabio inferior izquierdo asociada a un leve trismo y tumefacción. En la radiografía panorámica dental y en la tomografía mandibular se evidenció la presencia de un área osteolítica multiloculada de la hemimandíbula izquierda con compromiso del tercer molar inferior izquierdo (fig. 2). En junio del mismo año se extrajo la lesión, en la cual se evidenció una neoformación constituida por una proliferación de células fusiformes. En ese contexto, además, se apreciaron numerosos histiocitos de citoplasma espumoso, presentando el tejido buena vascularización, con eritrocitos extravasados y depósitos de fibrina focal, así como escasas microcalcificaciones en un fondo estromal colagenoso (fig. 3A), resultando como diagnóstico fibroma fibrohistiocitario. Sin embargo, el paciente continuó con los síntomas iniciales, por lo que se realizó otra radiografía, que reveló persistencia de la lesión. En septiembre del mismo año se realizó una nueva cirugía de la neoformación que se encontraba adherida al nervio alveolar inferior homolateral con coronoidectomia homolateral, apreciándose en la segunda radiografía panorámica dental una amplia área osteolítica multiloculada con márgenes mal definidos de la rama y ángulo mandibular izquierdo.

 Primera biopsia: se observa una proliferación celular en la que se puede apreciar que algunas células presentan un citoplasma lipoídico mientras que otras son redondas y fusiformes (H&E, 100×). Las microcalcificaciones se encuentran en un contexto colagenoso (recuadro). B) Segunda biopsia: se observa un aumento en el número de células; la mayoría son redondas y fusiformes, mientras que algunas presentan un citoplasma claro (H&E, 200×). C) Inmunotinción para CD68 en la que se observan positivos los elementos histiocitarios (200×). D) Inmunotinción para S100 en la que se observa negatividad en los elementos histiocitarios (200×). E) Inmunotinción para actina en la que se observan positivos los elementos estromales, mientras que los histiocitos permanecen negativos (400×).")
A) Primera biopsia: se observa una proliferación celular en la que se puede apreciar que algunas células presentan un citoplasma lipoídico mientras que otras son redondas y fusiformes (H&E, 100×). Las microcalcificaciones se encuentran en un contexto colagenoso (recuadro). B) Segunda biopsia: se observa un aumento en el número de células; la mayoría son redondas y fusiformes, mientras que algunas presentan un citoplasma claro (H&E, 200×). C) Inmunotinción para CD68 en la que se observan positivos los elementos histiocitarios (200×). D) Inmunotinción para S100 en la que se observa negatividad en los elementos histiocitarios (200×). E) Inmunotinción para actina en la que se observan positivos los elementos estromales, mientras que los histiocitos permanecen negativos (400×).
A nivel de la segunda biopsia se observó una neoformación constituida por una proliferación de células de tamaño medio, con citoplasma claro, algunas fusiformes, con un fondo estromal colagenoso (fig. 3B). Focalmente se apreciaron microcalcificaciones intersticiales y un infiltrado inflamatorio linfohistiocitario. Con la tinción de Ziehl-Nielsen no se evidenciaron micobacterias. El estudio de inmunohistoquímica fue positivo focalmente en los elementos proliferantes para CD68 (fig. 3C) y negativo para el S100, actina (fig. 3D,E), CD1a, CD31 y CD34.
Al sospechar que se trataba de una enfermedad de Erdheim-Chester, se envió este caso a consulta al Centro Nacional de Tumores de Tejidos Blandos (Dr. A. P. Dei Tos), donde, utilizando la radiografía junto con la biopsia, se diagnosticó como posible enfermedad de Erdheim-Chester.
DiscusiónLa enfermedad de Erdheim-Chester es una forma inusual de proliferación anormal de histiocitos no-Langerhans descrita por primera vez por William Chester en 19301,2 y de etiología desconocida. Histológicamente esta enfermedad incluye macrófagos, células gigantes aisladas, infiltrado inflamatorio crónico con predominio de linfocitos, escasos o aislados eosinófilos y zonas de fibrosis. Los núcleos de los macrófagos habitualmente no muestran barras cromatínicas ni seudoinclusiones3. Los estudios sobre el origen monoclonal de la proliferación han llegado a conclusiones contrapuestas, rehusadas por algunos investigadores4 y confirmadas por otros5.
En la presente revisión presentamos 2 casos en los que, como datos comunes, se observaron compromiso del maxilar y dolor óseo. Ambos pacientes fueron de sexo masculino, y sus edades fueron de 61 y 22 años, respectivamente, lo cual coincide con lo expuesto en la literatura2.
La presentación característica radiográfica es la osteosclerosis simétrica de huesos largos. El hallazgo más importante, que solo se observó en uno de nuestros casos, es la presencia de masas alrededor de varios órganos que los infiltran y encapsulan. Los pacientes presentan dolor óseo, sobre todo a nivel de las extremidades inferiores, y frecuentemente déficits neurológicos, exoftalmos indoloros bilaterales, enfermedad extraesquelética y diabetes insípida2,3,6, como se observó en nuestro primer caso.
La fibrosis de la aorta abdominal, según la literatura, se puede observar con escáner o RMN hasta en el 56% de los pacientes, llegando a infiltrar el espacio retroperitoneal y los uréteres7-9, como se observó en el primer caso. La presencia de insuficiencia cardiaca es un hallazgo común4, y en esta oportunidad llegó a evolucionar como insuficiencia mitral.
Desde el punto de vista histopatológico, en el primero de nuestros casos se realizó biopsia de médula ósea en la que se observaron agregados de células grandes de aspecto histiocitario con núcleos blandos, centrales y citoplasma espumoso S100 y CD1a negativo y CD68 positivo, lo cual nos condujo al diagnóstico de histiocitosis de células no-Langerhans (enfermedad de Erdheim-Chester).
En nuestro primer caso, además de los marcadores previamente descritos, también se observó positividad para el PGM1, un anticuerpo monoclonal que reconoce el antígeno CD68. El PGM1 ha mostrado una mayor especificidad para reconocer células del linaje monocítico/macrofágico en comparación con el anti CD68, que puede expresarse en otros tipos celulares10.
En el diagnóstico diferencial debemos tener en cuenta todas las enfermedades que cursan con agregados tisulares de histiocitos no-Langerhans, que representan un largo listado de diferentes entidades, como fue detallado por Weitzman y Jaffe11. Estos investigadores clasificaron las histiocitosis no-Langerhans en cutáneas y sistémicas, con la enfermedad de Erdheim-Chester perteneciente a la segunda categoría, subrayando la importancia de la clínica, así como la edad de inicio, la evolución y los estudios radiográficos. En nuestros casos, el primer paciente tuvo una larga historia de compromiso multisistémico y también la característica fibrosis retroperitoneal. En el segundo paciente, su cuadro está enfocado a la afección maxilar.
Aunque en la literatura se han reportado pacientes que presentan histiocitosis de células de Langerhans (S100+, CD1a+, CD68–) asociada con la enfermedad de Erdheim-Chester, en nuestros casos esto no se evidenció5-12. Igualmente, en los casos de histiocitos no-Langerhans S100 y CD68 positivos y CD1a negativos se debe considerar la enfermedad de Rosai-Dorfman. Los histiocitos de esta enfermedad son muy característicos, ya que contienen en su citoplasma células inflamatorias, principalmente linfocitos (emperipolesis). Aunque la emperipolesis no sea exclusiva de la enfermedad de Rosai-Dorfman, su ocurrencia en histiocitos S100 positivos se considera diagnóstica de esta enfermedad.
Sin embargo, llegar a la conclusión diagnóstica de enfermedad de Erdheim-Chester no siempre es tan característico, ya que en el segundo caso el diagnóstico emitido en la biopsia fue el de fibroma fibrohistiocitario, y aunque los elementos histiocitarios fueron positivos para CD68 y negativos para S100, se llegó a esta conclusión debido a que el tejido presentó también inmunopositividad para actina en el estroma. Pero después de este diagnóstico la lesión recurrió, por lo que se comenzó a pensar en esta enfermedad, y aunque actualmente este paciente aún no ha presentado compromiso de ningún otro órgano, se recomienda un estrecho seguimiento médico.
Las opciones terapéuticas actuales incluyen esteroides, alcaloides, antraciclinas y, más recientemente, interferón alfa. En nuestro primer paciente se utilizó colchicina para el manejo de la fibrosis, pero después de que fuera diagnosticado con enfermedad de Erdheim-Chester su tratamiento cambió a interferón alfa13, sin resultados positivos, por lo que luego fue sustituido por imatinib14. Este último es un inhibidor de la tirosina cinasa, y promete ser un nuevo tratamiento para pacientes con formas que no han tenido buena respuesta con el interferón alfa.
Ya que la enfermedad de Erdheim-Chester presenta hallazgos característicos que pueden afectar tanto el tejido óseo como los órganos, en el momento de diagnosticar esta entidad se deben revisar con suma cautela las secciones histológicas, especialmente en biopsias que presentan patologías fibromatosas en localizaciones poco comunes, considerando que en el caso número 2 solo se pensó en esta enfermedad en el momento en que el fibroma recidivó. Por otro lado, los huesos craneofaciales están frecuentemente involucrados en la enfermedad de Erdheim-Chester, en la cual también se encuentra descrita como única localización ósea15.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

