







El carcinoma de pulmón de no célula pequeña (CPNCP) presenta el mayor número de dianas terapéuticas identificadas, algunas de ellas con utilidad terapéutica. En la actualidad se considera imprescindible en estos pacientes determinar las mutaciones de EGFR, BRAF, KRAS y MET, las traslocaciones de ALK, ROS1, NTRK y RET y la expresión de PD-L1. El uso de la secuenciación masiva (next-generation sequencing [NGS]) facilita el diagnóstico molecular de forma precisa y permite determinar otras mutaciones emergentes, como la mutación de HER2 y los biomarcadores predictivos de respuesta a inmunoterapia.
En este consenso, un grupo de expertos en el diagnóstico y tratamiento del CPNCP seleccionado por la Sociedad Española de Anatomía Patológica (SEAP) y la Sociedad Española de Oncología Médica (SEOM) ha evaluado la información actualmente disponible y propone una serie de recomendaciones para optimizar la determinación y utilización en la práctica clínica diaria de los biomarcadores.
Non-small cell lung cancer (NSCLC) presents the greatest number of identified therapeutic targets, some of which have therapeutic utility. Currently, detecting EGFR, BRAF, KRAS and MET mutations, ALK, ROS1, NTRK and RET translocations, and PD-L1 expression in these patients is considered essential. The use of next-generation sequencing (NGS) facilitates precise molecular diagnosis and allows the detection of other emerging mutations, such as the HER2 mutation and predictive biomarkers for immunotherapy responses.
In this consensus, a group of experts in the diagnosis and treatment of NSCLC selected by the Spanish Society of Pathology (SEAP) and the Spanish Society of Medical Oncology (SEOM) have evaluated currently available information and propose a series of recommendations to optimize the detection and use of biomarkers in daily clinical practice.
Los carcinomas de pulmón no célula pequeña (CPNCP) son el grupo de tumores con mayor número de dianas terapéuticas identificadas, algunas de las cuales tienen utilidad clínica desde los estadios más precoces. Sin duda, el diagnóstico molecular correcto es una obligación para ofrecer la mejor opción terapéutica a cada paciente, y debe aplicarse lo más ampliamente posible. Afortunadamente, en los últimos años se han logrado avances importantes tanto en técnicas diagnósticas moleculares como en terapias personalizadas. Este documento pretende ofrecer nuevas recomendaciones para la determinación de biomarcadores predictivos en CPNCP, y será una actualización de las ya publicadas en 2012, 2015 y 2020, fruto de este consenso entre la Sociedad Española de Oncología Médica (SEOM) y la Sociedad Española de Anatomía Patológica (SEAP)1.
Requisitos para el análisis de una muestra biológica óptimaExisten varios tipos de muestras que pueden resultar útiles para el estudio de biomarcadores: biopsias, piezas quirúrgicas o citologías, siempre que tengan una cantidad suficiente de células tumorales y hayan sido correctamente procesadas2,3. La decisión acerca de cuál considerar dependerá de la experiencia y de las tecnologías disponibles en cada laboratorio. Se recomienda en general usar la muestra más reciente, sobre todo en los pacientes previamente tratados4.
Una muestra debe conservarse en formol neutro tamponado al 10% de seis a 48 h en función de su tamaño (6-12 h en muestras pequeñas y 24-48 h en resecciones quirúrgicas), con la presencia de un mínimo de 50-100 células viables para los estudios de inmunohistoquímica (IHQ) y de hibridación fluorescente in situ (fluorescence in situ hybridization [FISH]). Se recomienda evitar el uso de fijadores alternativos, como fijadores mercuriales o alcohólicos. En el caso de las muestras citológicas, el bloque celular se procesa exactamente igual que la biopsia. Las extensiones se fijan en alcohol de 96° y se recomienda teñirlas con Papanicolaou. A partir de esos materiales también se pueden realizar la mayoría de los estudios de biomarcadores5. Para las técnicas basadas en la extracción de ácidos nucleicos, se debe conocer el umbral del límite de detección (LDD) del método utilizado. Cada tipo de técnica tiene unos requisitos mínimos diferentes, oscilando entre 30% de células tumorales para la secuenciación directa, 5% para la reacción en cadena de la polimerasa (polymerase chain reaction [PCR]) en tiempo real o 20% para la secuenciación de nueva generación (next-generation sequencing [NGS])6. Además, el tipo de mutaciones puede cambiar el umbral de sensibilidad. Así, se requiere un rango de contenido de ácido nucleico de entre 5 y 10% para detectar mutaciones puntuales y pequeñas inserciones o deleciones, y hasta 30% para analizar correctamente alteraciones en el número de copias6. Se recomienda disponer de dos métodos alternativos para realizar una determinación molecular redundante, si fuera necesario.
Respecto al manejo de todo tipo de muestras biológicas, es preciso utilizar protocolos de aprovechamiento que permitan tanto el diagnóstico anatomopatológico como la detección de biomarcadores.
¿Qué biomarcadores deberían analizarse en el CPNCP?En la tabla 1 se describen los biomarcadores que se deben determinar de forma obligada en pacientes con CPNCP y en la tabla 2 se describen otros biomarcadores de interés en estos pacientes.
Biomarcadores esenciales en pacientes con CPNCP
| Gen/proteína | Alteración predictiva | Metodología |
|---|---|---|
| EGFR | Mutación | PCR: secuenciación de sanger, PCR en tiempo real y NGS |
| ALK | Reordenamiento | IHQ, FISH, PCR en tiempo real y NGS |
| ROS1 | Reordenamiento | IHQ (cribado), FISH, PCR en tiempo real y NGS |
| BRAF V600 | Mutación | PCR en tiempo real y NGS |
| PD-L1 | Sobreexpresión | IHQ |
| NTRK | Reordenamiento | IHQ (cribado), PCR en tiempo real y NGS |
| RET | Reordenamiento | FISH, PCR en tiempo real y NGS |
| KRAS | Mutación | PCR: secuenciación de sanger, PCR en tiempo real y NGS |
| MET | MutaciónAmplificación | NGSFISH, PCR en tiempo real y NGS |
ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; EGFR: epidermal growth factor receptor; FISH: hibridación fluorescente in situ; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MET: mesenchymal epithelial transitionfactor; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; RET: rearranged during transfection; ROS1: c-ros oncogene 1.
Otros biomarcadores de interés en pacientes con CPNCP
| Gen/proteína | Alteración predictiva | Metodología |
|---|---|---|
| HER2 | MutaciónAmplificación | NGSFISH, PCR en tiempo real, NGS |
| TMB | Mutaciones | NGS |
| STK11 | Mutación | NGS |
| KEAP1 | Mutación | NGS |
| MSI | Patrón de hipermutación | IHQ, PCR, NGS |
FISH: hibridación fluorescente in situ; HER2: human epidermal growth factor receptor 2; IHQ: immunohistoquímica; KEAP1: Kelch-like ECH-associated protein 1; MSI: microsatellite instability-high; NGS: next-generation sequencing; PCR: polymerase chain reaction; STK11: serine/threonine kinase 11; TMB: tumour mutation burden.
Las mutaciones en el gen del receptor del crecimiento epidermico (epidermal growth factor receptor [EGFR]) se identifican en aproximadamente 10-16% de los CPNCP, siendo más frecuentes en adenocarcinomas y en pacientes no fumadores7. Las mutaciones más frecuentes y que se relacionan directamente con la sensibilidad a inhibidores de tirosin-quinasa (tyrosine kinase inhibitor [TKI]) anti-EGFR afectan al exón 19, y consisten en deleciones que conservan el marco de lectura (in-frame deletions) entre los codones 746 y 759 (aminoácidos leucina, arginina, glutamato y alanina, LREA) (45-50%), seguidas de mutaciones puntuales tipo «cambio de sentido» (missense point mutations) en el exón 21 al sustituir el aminoácido leucina por arginina en la posición 858 (L858R) (35-45%). Hay varios EGFR-TKI aprobados para el tratamiento de primera línea de pacientes con enfermedad metastásica y mutaciones activadoras de EGFR (delección exón 19, L858R)8, entre ellos, osimertinib (opción preferida en la mayoría de las guías), gefitinib, erlotinib, afatinib y dacomitinib. Osimertinib está también aprobado como tratamiento adyuvante tras una resección quirúrgica completa en pacientes adultos con mutación activadora de EGFR.
Otras mutaciones, como las inserciones en el exón 20, deben ser detectadas debido a sus diferentes efectos y requisitos de tratamiento9.
En relación con la metodología, las pruebas clínicas para la determinación de EGFR deben ser capaces de detectar todas las mutaciones individuales que han sido informadas con una frecuencia de al menos 1% en los CPNCP EGFR mutados. Se recomienda utilizar métodos de alta sensibilidad. En cuanto a los informes de resultados, se deben especificar las mutaciones que han sido detectadas y la sensibilidad de los métodos de detección utilizados, entre otros datos3.
Las recomendaciones iniciales de diagnóstico de mutaciones en EGFR han sufrido algunos cambios, entre los que destaca el hecho de que cualquier muestra citológica con celularidad y preservación adecuada puede ser utilizada, la necesidad de utilizar técnicas con elevada sensibilidad en comparación con la secuenciación tipo Sanger como método de referencia y la falta de sensibilidad de la IHQ para el diagnóstico de mutaciones en la práctica clínica7.
La mayoría de los pacientes con mutaciones de EGFR sensibilizadoras (deleción exón 19 y mutaciones exón 21, L858R) reciben un TKI anti-EGFR, siendo el mecanismo molecular de resistencia adquirida más frecuente la mutación EGFR T790 M en pacientes que reciben un TKI anti-EGFR de primera o segunda generación (50-60% de los casos). Para la determinación de esta mutación, se deben utilizar técnicas que sean capaces de detectarla en al menos 5% de células viables, incluyendo los nuevos métodos de PCR digital10.
Los mecanismos que conducen a la resistencia adquirida contra los TKI varían e incluyen mutaciones intragénicas, amplificación o fusión génica y adaptación funcional con transformación histológica. Por consiguiente, los mecanimos de resistencia adquirida deben monitorizarse mediante biopsia tumoral o biopsia líquida (BL)11.
ALKLos reordenamientos de la quinasa del linfoma anaplásico (anaplastic lymphoma kinase [ALK]) están presentes en 2-5% de los CPNCP avanzados9. Estos tumores surgen con mayor frecuencia en pacientes jóvenes y en mujeres con o sin una mínima exposición previa al tabaquismo. La enfermedad suele ser agresiva en su curso clínico y se presenta con eventos tromboembólicos, siendo comunes las metástasis en el hígado, las superficies serosas y el cerebro12. Los resultados, incluyendo la supervivencia, mejoran notablemente con los TKI específicos de ALK y, en la actualidad, la mediana de supervivencia global de los pacientes en estadio IV suele superar los cinco años. Crizotinib fue el primer fármaco aprobado en este contexto y, desde entonces, los TKI de segunda (ceritinib, alectinib y brigatinib) y tercera generación (lorlatinib) están disponibles en la Unión Europea para el tratamiento de pacientes no tratados y para aquellos que siguen una progresión con inhibidores previos13. El beneficio de los fármacos individuales en estos pacientes pretratados depende del mecanismo de resistencia, que con frecuencia implica mutaciones adquiridas en la quinasa ALK14.
Los tipos de histología elegibles para las pruebas de reordenamiento de ALK deben incluir todos los adenocarcinomas, carcinomas con evidencia histológica no escamosa y tumores escamosos en pacientes menores de 50 años o con poca o ninguna exposición al tabaco (es decir, < 10 paquetes al año)15. Los métodos clave para detectar el reordenamiento del gen ALK son IHQ, FISH y NGS. En la actualidad, la IHQ representa un método rápido, fiable y rentable para detectar fusiones ALK16. Su uso en frotis citológico es bastante controvertido, aunque estudios recientes han demostrado la idoneidad del método5. Los anticuerpos más utilizados para la detección de reordenamientos son D5F3 (Ventana® ALK [D5F3] CDx Assay, Tucson, Arizona, EE. UU.) y 5A4 (Novocastra®, Leica Biosystems®, Buffalo Grove, Illinois, EE. UU.), aunque este último no está incluido en kit de diagnóstico17. El apéndice cecal es adecuado tanto como control positivo como negativo. Debe fijarse y procesarse en las mismas condiciones que la muestra del paciente. Un caso de tumor positivo también se puede utilizar como control.
El papel de la FISH como metodología estándar óptima es actualmente controvertido, aunque existen algoritmos de lectura automatizados aprobados por la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) que aumentan considerablemente su fiabilidad18. Cuando hay un resultado positivo de IHQ por una fuerte tinción citoplásmica granular con cualquiera de los anticuerpos 5A4 o D5F3, la confirmación por una segunda técnica no es obligatoria15. Sin embargo, es muy recomendable en casos que no son concluyentes. Esta redundancia diagnóstica también es útil si se encuentra una tinción FISH inusual19.
Por último, los métodos basados en NGS y ensayos de ARN son muy específicos y existen numerosos estudios que demuestran su valor para detectar fusiones en pacientes que muestran resultados negativos con otras técnicas19. Las pruebas de variantes para reordenamientos específicos en ALK, que pueden proporcionar información útil en términos de predicción de la respuesta a inhibidores específicos, aún no tienen datos suficientes para su recomendación, aunque podrían ser útiles en el futuro9,14,19. En algunas circunstancias, la biopsia líquida puede reemplazar el análisis de biomarcadores tumorales tisulares, y el perfil ALK en el ADN tumoral circulante (ctADN) puede servir como una herramienta de orientación del tratamiento20,21.
Las mutaciones de ALK están emergiendo como importantes mecanismos de resistencia a los TKI de ALK, y las pruebas de mutación de ALK en este escenario pueden proporcionar información crucial para guiar el tratamiento, ya que los TKI de ALK de nueva generación muestran diferentes eficacias contra diferentes mutaciones de ALK22.
ROS1El oncogén c-ros 1 (c-ros oncogene 1 [ROS1]) codifica un receptor con actividad de tirosina quinasa. Los reordenamientos de genes activadores con varios genes asociados se encuentran en aproximadamente 1% de los CPNCP, en particular los que surgen en pacientes jóvenes no fumadores23. Estos tumores se asocian frecuentemente con eventos trombóticos y tienen propensión a desarrollar metástasis en el sistema nervioso central (SNC)24. Las fusiones de ROS1 ocurren casi exclusivamente en adenocarcinomas, frecuentemente en aquellos con un componente sólido y células en anillo de sello25. Este perfil histológico también es típico de los tumores que albergan una translocación de ALK. De hecho, ambos receptores tienen una similitud de 77% en su dominio de unión al ATP.
Crizotinib fue el TKI inicial aprobado para el tratamiento de primera o segunda línea de pacientes con cáncer de pulmón en estadio IV con reordenamiento de ROS126. Más recientemente se están estudiando TKI como lorlatinib, entrectinib y repotrectinib, pero aún no están aprobados para esta indicación27.
Actualmente se recomienda realizar pruebas de ROS1 en pacientes con adenocarcinoma de pulmón en estadio avanzado, independientemente de las características clínicas. Las pruebas de ROS1 no se recomiendan en el carcinoma de células escamosas, excepto en fumadores de grado bajo o leve27. Se utilizan tres técnicas para detectar reordenamientos de ROS1: IHQ, técnicas citogenéticas, particularmente FISH9, y técnicas moleculares como PCR con transcripción inversa (reverse transcription PCR [RT-PCR]) y particularmente NGS15,19. Generalmente se recomienda IHQ como método de detección y los casos positivos deben confirmarse con otro método ortogonal (por ejemplo, FISH o NGS), debido a la especificidad variable de los dos anticuerpos disponibles comercialmente (D4D6, Cell Signaling Technology y SP384, Ventana Medical Systems®)3,15,28. La muestra para analizar debe incluir al menos 20 células tumorales y cada laboratorio debe validar su propio rango de interpretación15,19,28. Debe disponerse de un control externo, distinto del apéndice cecal, y es recomendable tener también un control tumoral positivo. También se ha considerado como control la existencia de neumocitos reactivos peritumorales positivos. Cabe destacar que la expresión de ROS1, típicamente focal, se puede encontrar en hasta un tercio de los tumores sin reordenamientos subyacentes de ROS1, pero con otras alteraciones genómicas (por ejemplo, mutaciones de EGFR, virus del sarcoma de rata Kirsten (kirsten rat sarcoma virus [KRAS]), BRAF o receptor 2 del factor de crecimiento epidérmico humano (human epidermal growth factor receptor 2 [HER2]) y reordenamientos de ALK28,29. Además, también se ha observado inmunotinción no específica en el subtipo histológico de adenocarcinoma mucinoso infiltrante y en tejido no tumoral30.
FISH es una de las técnicas de referencia. Utiliza sondas separables de dos colores y recomienda un recuento de al menos 50 células tumorales15,28,30,31. Un tumor debe considerarse positivo cuando al menos 50% de las células tumorales tienen señales de rotura (separadas por ≥ 1 diámetro de señal) o señales aisladas de 3’ (frecuentemente marcadas con fluorocromo verde)30. Se han descrito falsos positivos y falsos negativos, atribuibles tanto a causas metodológicas como biológicas30,32. Por último, las tecnologías NGS (basadas en ADN o ARN) han mostrado una alta sensibilidad y especificidad en muestras tumorales y también en ctADN20,21.
BRAFLas mutaciones del protooncogén B-Raf (B-Raf proto-oncogene [BRAF]) se observan en 2% de los carcinomas de pulmón, son excluyentes con otras y aparecen en su mayoría en adenocarcinomas, sobre todo de tipo papilar (80%)33. La mutación más frecuente es la BRAFV600E (Val600Glu) (50%), predomina en mujeres y puede implicar una mayor agresividad tumoral, mientras que el resto son más habituales en varones o pacientes con hábito tabáquico34. La Agencia Europea del Medicamento (EMA) y la FDA han aprobado dabrafenib y trametinib tras la eficacia demostrada en los ensayos clínicos fase 2 en pacientes con mutación BRAF V6009. En el caso de la FDA, la aprobación incluye la necesidad de determinar la mutación con el panel de NGS Oncomine Dx Target Test®35.
Actualmente, se permite cualquier método de PCR con sensibilidad y calidad adecuadas para identificar mutaciones BRAF. Sin embargo, no se recomienda llevar a cabo la determinación de esta mutación de forma individual, por lo que se suele estudiar en paneles de NGS, que incluyan como mínimo el análisis de los exones 11 y 15 de ese gen.
PD-L1Los inhibidores del punto de control inmunitario (ICI), los inhibidores del receptor de la proteína programada-1/ligando 1 de muerte celular programada (programmed cell death protein-1/ligand-1 [PD-1/PD-L1]) y, en menor medida, los bloqueadores de la proteína 4 asociada a los linfocitos T citotóxicos (CTLA-4), han demostrado ser una estrategia eficaz en el tratamiento del cáncer de pulmón, CPNCP y cáncer de pulmón de células pequeñas (CPCP), en los últimos 15 años36. En la actualidad, los datos de ensayos clínicos aleatorizados los respaldan como el tratamiento estándar para pacientes con CPNCP localmente avanzado o metastásico, ya sea en monoterapia o en combinación con quimioterapia37. Más recientemente, el bloqueo de PD-1/PD-L1 también ha demostrado ser eficaz en el contexto adyuvante y neoadyuvante en pacientes con enfermedad temprana38,39. Aunque PD-L1 está lejos de ser un biomarcador ideal, la magnitud del beneficio de los bloqueadores de PD-1/PD-L1 en monoterapia está relacionada con la expresión tumoral de PD-L138. Por el contrario, la expresión de PD-L1 no predice la eficacia de los regímenes combinados de quimioterapia con inhibidores de PD-1/PD-L1, PD-1 con bloqueadores de CTLA-4 o quimioterapia con PD-1 y bloqueadores de CTLA-437.
La prueba de PD-L1 se basa en IHQ, actualmente es la única prueba predictiva validada. La diversidad de ensayos IHQ y puntos de corte que definen un resultado positivo ha sido una fuente de confusión y ha impulsado una serie de esfuerzos de armonización por parte de la comunidad científica40,41. Las guías actuales para la determinación del biomarcador PD-L1 recomiendan las condiciones preanalíticas habituales de las pruebas IHQ. La expresión de PD-L1 se evalúa determinando el porcentaje de células tumorales con tinción parcial o total de la membrana de cualquier intensidad.
Hay varios clones de PD-L1 disponibles para pruebas IHQ. Los cuatro más utilizados en los laboratorios de patología son 22C3 y 28-8 de Agilent (que comparten la plataforma de diagnóstico Autostainer LINK 48® de Agilent®), SP263 de MedImmune®/Ventana® y SP142 de Spring®/Bioscience®/Ventana® (que comparten la plataforma de diagnóstico Ventana® BenchMark)1. Las características de rendimiento de los ensayos 22C3 y 28-8 parecen ser similares según la evaluación en paralelo en cohortes retrospectivas. SP263 y E1L3N, utilizados en la práctica habitual pero no aprobados como pruebas de diagnóstico complementarias, pueden mostrar patrones de tinción comparables a los de los ensayos aprobados cuando se validan correctamente. El único valor atípico consistente ha sido el ensayo SP142, que muestra una menor tinción de células tumorales, a pesar de que el anticuerpo SP142 reconoce epítopos idénticos o casi idénticos como SP263 y E1L3N42. El ensayo SP142 se optimizó tanto para la puntuación de células tumorales como de células inmunitarias. Sin embargo, su desempeño como marcador de células inmunitarias se confunde aún más por el escaso acuerdo entre observadores en la interpretación de la expresión de células inmunitarias43.
En cuanto a la selección de muestras, si hay más de un bloque de tejido disponible para un tumor determinado, se debe analizar la muestra más representativa. Se puede analizar más de un bloque cuando el patólogo determina que se necesitan pruebas adicionales para establecer el estado de PD-L1 del tumor. Si se analizan bloques adicionales de la misma muestra, los resultados de todos los bloques analizados deben combinarse como si estuvieran presentes en un solo bloque de parafina44.
No es raro que el único material disponible provenga de muestras citológicas. En estos casos, se debe tener en cuenta que el uso de kits IHQ PD-L1 que estén validados para muestras de biopsia fijadas en formalina e incluidas en parafina (formalin-fixed paraffin-embedded [FFPE]), y no específicamente para muestras citológicas, pueden ser usados si las muestras citológicas se procesaron de acuerdo con las mismas condiciones preanalíticas que requieren los kits45.
NTRKLas fusiones del receptor tirosin-kinasa neurotrófico (neurotrophic tyrosine receptor kinase [NTRK]) pueden estar presentes en una gran variedad de tumores, tanto en adultos como en pacientes de edad pediátrica, siendo la frecuencia estimada en CPNCP menor de 1%. La mayoría se encuentran en adenocarcinomas y el gen más frecuentemente implicado es NTRK19.
No existen datos clínicos o patológicos característicos de estos pacientes, pero es crucial identificarlos puesto que se dispone de inhibidores del receptor de tropomiosina quinasa (iTRK) como larotrectinib y entrectinib, que han sido aprobados por la EMA y la FDA para el tratamiento de tumores con fusión de NTRK46. A pesar de la marcada eficacia, el desarrollo de resistencias es frecuente, existiendo ya resultados clínicos con iTRK de segunda generación47.
Para la detección de estas alteraciones se recomiendan dos estrategias: mediante NGS con un panel que incluya el estudio de los tres genes (es decir, NTRK1, 2 y 3) y un adecuado número de parejas de reordenamiento o un cribado mediante IHQ con confirmación posterior obligatoria de todos los resultados positivos obtenidos mediante NGS48,49.
RETLas fusiones del gen reorganizado durante la transfección (rearranged during transfection [RET]) se observan en distintos tipos de tumores, siendo su frecuencia en CPNCP de 1-2%, principalmente en adenocarcinomas de pacientes no fumadores. KIF5B es la pareja de reordenamiento más frecuente50. La presencia de calcificaciones en forma de cuerpos de psamoma debe sugerir fusiones RET51.
Actualmente se dispone de inhibidores selectivos, como selpercatinib y pralsetinib con tasas de respuesta elevadas, aunque su aprobación por las agencias reguladoras para el tratamiento de primera línea en pacientes con enfermedad avanzada está condicionada a los resultados que se obtengan en los estudios fase III en marcha52,53.
El método de detección óptimo de la fusión RET es la NGS, pero también se pueden utilizar FISH o PCR54. Para diseñar un algoritmo eficiente de búsqueda de fusiones en RET, es importante considerar que: i) la NGS basada en el estudio del ARN es más sensible que si solo se investiga el ADN, y ii) los resultados del FISH pueden ser difíciles de interpretar55,56.
KRASLas mutaciones en KRAS se identifican en 25% de los pacientes con CPNCP. Se encuentran en todos los subtipos histológicos de adenocarcinoma, aunque son más habituales en la variante mucinosa invasiva. También se detectan en 5% de los tumores escamosos57. Su presencia confiere heterogeneidad biológica y clínica, y puede no tener valor pronóstico58.
Las mutaciones en KRAS se suelen localizar en los codones 12, 13 y 61. Las del codón 12 suponen 80% de casos, y generalmente son transversiones de glicina por cisteína (KRASG12C), valina (KRASG12V), o aspártico (KRASG12D), siendo su frecuencia de 10-13, 5 y 4%, respectivamente59. Las mutaciones KRASG12C y KRASG12V se suelen relacionar con el tabaquismo y activan la vía RalGDS/Ral/FLIP, mientras que la mutación KRASG12D es más típica de pacientes no fumadores y parece activar las vías PI3K/AKT/mTOR y RAF/MEK/ERK. Además, KRASG12C muestra mayor fosforilación de ERK1/2.
Más de 50% de CPNCP con mutaciones KRAS presentan otra mutación, y se pueden establecer tres subgrupos: el subgrupo KP tiene mutaciones en la proteína tumoral 53 (tumour protein 53 [TP53]) y supone 40% de los casos; el subgrupo KL donde se identifican serina/treonina quinasa 11 (serine/threonine kinase 11 [STK11]), proteína asociada a ECH de tipo Kelch 1 (Kelch-like ECH-associated protein 1 [KEAP1]) o quinasa hepática B1 (liver kinase B1 [LKB1]) y suele asociarse con bajos porcentajes de PD-L1; y el subgrupo KC caracterizado por la inactivación de CDK2A/B y se asocia con histología mucinosa60,61. Por el contrario, es muy raro encontrar mutaciones de EGFR, por lo que las mutaciones de KRAS y EGFR se consideran mutuamente excluyentes.
La técnica utilizada para identificar mutaciones KRAS suele ser la PCR. Hasta el momento, no se recomienda su determinación de forma aislada, pero debería incluirse en los paneles de NGS62.
Tras años sin terapias eficaces, se han desarrollado varios inhibidores que han demostrado actividad en ensayos fase II frente a la mutación KRASG12C, como sotorasib y adagrasib63,64; por lo que han sido aprobados por la FDA, aunque el beneficio de estos agentes en monoterapia o en combinación, y a qué pacientes se les debe administrar, se están estudiando en ensayos de fase III.
METLa activación oncogénica del gen del factor de transición epitelial mesenquimal (mesenchymal epithelial transition factor [MET]) en el CPNCP puede ocurrir principalmente por amplificación (1-5%) o por la presencia de mutaciones en el exón 14 (3-4%) que reducen la degradación de la proteína MET9. En los CPNCP de morfología sarcomatoide, la frecuencia de mutaciones en el exón 14 puede llegar a ser de 22%65. Entre 5-20% de los pacientes con mutaciones en EGFR adquieren resistencia a EGFR-TKI a través de la amplificación de MET9.
Actualmente, los resultados de varios ensayos clínicos demuestran la actividad y tolerabilidad de los fármacos orales como capmatinib, tepotinib y savolitinb en pacientes con mutaciones en el exón 14 de los cuales capmatinib y tepotinib ya han sido aprobados por la EMA9. También se están estudiando anticuerpos conjugados, como telisotuzumab vedotin, anticuerpos biespecificos como amivantamab y otros en pacientes con amplificación de MET.
La técnica de elección para estudiar la amplificación de MET es FISH, ya que permite estimar con más precisión el aumento en el número de copias y la amplificación clonal. Debido a la heterogeneidad de las mutaciones en el exón 14, el método de detección óptimo en este caso es la NGS. Se debe utilizar un panel de NGS con suficiente cobertura. Para evitar falsos negativos, se recomienda utilizar un panel de ARN9. Se puede detectar la sobreexpresión de MET por la amplificación o las mutaciones del gen, pero el valor predictivo de la IHQ de MET es todavía controvertido7,9.
HER2En los CPNCP se puede encontrar sobreexpresión, amplificación y mutaciones de HER2, identificándose en 3-38, 3 y 1-4%, respectivamente66. Las mutaciones más frecuentes son inserciones del exón 20 (dominio tirosina quinasa), con la inserción/duplicación de los cuatro aminoácidos tirosina, valina, metionina y alanina (YVMA) en el codón 776 (YVMA 776-779 ins) la más frecuente (80-90%)66,67. Estas mutaciones se asocian sobre todo con adenocarcinomas, pacientes no fumadores y mujeres68. Los datos más recientes apuntan a que estas mutaciones son los mejores predictores de beneficio clínico a terapias anti-HER2 (por ejemplo, trastuzumab deruxtecan), independientemente del tipo de mutación y de la presencia de sobreexpresión y amplificación67.
En relación con las metodologías para evaluar el estado de HER2, la NGS basada en ADN o ARN es el método más apropiado para seleccionar a los pacientes, frente a la IHQ y el FISH67. La amplificación se ha descrito como un mecanismo de resistencia tras los tratamientos dirigidos66.
Biomarcadores inmunitarios con valor potencialLa carga mutacional tumoral (tumour mutation burden [TMB]) se refiere al número de mutaciones somáticas presentes en el tumor, excluyendo polimorfismos y mutaciones germinales de todas las variantes expresadas por megabase en el exoma estudiado. Las mutaciones adquiridas por las células tumorales pueden conducir a una estructura proteica anormal y, en consecuencia, a la expresión de neoantígenos que pueden provocar una respuesta inmune. Curiosamente, no existe una correlación clara entre la expresión de PD-L1 y TMB69. Muchos estudios han demostrado que la TMB alta en tumores da como resultado un mejor efecto terapéutico con la inmunoterapia anti-PD-1/PD-L1, incluso en algunos cánceres de pulmón69, pero no existe una validación definitiva para el uso de esta inmunoterapia en la práctica clínica. Sin embargo, el análisis exploratorio del ensayo Keynote 042 sugiere que entre los pacientes con tumores que expresan PD-L1 en ≥ 50% de las células, solo aquellos cuyo TMB fue superior a la mediana exhibieron algún beneficio terapéutico con los inhibidores de PD-1/PD-L1 en comparación a la quimioterapia70. De hecho, la TMB no es un predictor confiable del resultado para CPNCP o CPCP tratados con quimioterapia con bloqueo de ICI o regímenes de inmunooncología dual.
Con respecto a las pruebas de TMB, la NGS dirigida se considera una buena alternativa a la secuenciación masiva más compleja, y algunos datos recientes han validado el uso de paneles grandes71,72. Todavía se requieren estudios de armonización para validar la interconectividad entre diferentes estudios de NGS, la heterogeneidad del número de genes incluidos, la cobertura horizontal, la profundidad óptima requerida, el tipo de secuenciación química y los algoritmos bioinformáticos utilizados71. Si finalmente se aprueban medicamentos basados en los puntos de corte de TMB, los esfuerzos de armonización en curso podrían ser muy útiles. La detección de TMB en la sangre (blood TMB [bTMB]) es factible, pero aún faltan datos sólidos sobre su utilidad clínica.
Inestabilidad de microsatélites alta/deficiente reparación de MisMatch (microsatellite instability-high/deficient MisMatch repair [MSI-H/dMMR]) predice la eficacia de las ICI en el cáncer gástrico y el cáncer de colon. Sin embargo, la incidencia de MSI-H/dMMR en el cáncer de pulmón es baja73, y se necesita más investigación para determinar si MSI-H/dMMR se puede utilizar como biomarcador predictivo en este contexto. En la actualidad, la medida estándar comúnmente utilizada para valorar MSI-H es el método Bethesda74. Es de destacar que los pacientes con MSI-H tienen una mayor probabilidad de tener TMB alta, pero no al revés75.
También se ha investigado el papel predictivo de las aberraciones genómicas subyacentes al cáncer de pulmón76. Las alteraciones genéticas típicamente percibidas como asociadas a la respuesta inmune incluyen aquellas en TP53 o KRAS y, por el contrario, las aberraciones que afectan a EGFR, ALK, ROS, RET, KEAP1 o LKB1 tienen menos probabilidades de estar asociadas con el beneficio del bloqueo del punto de control61,76. En cualquier caso, en la actualidad, los datos disponibles no respaldan recomendaciones de tratamiento basadas únicamente en dichas determinaciones genómicas.
Los biomarcadores inflamatorios tumorales, como las firmas genéticas relacionadas o el contenido de células tisulares (subtipos de células T, células mieloides, etc.) están en fase de investigación en la actualidad, ya que son inmunoterapia relacionada con la sangre periférica y biomarcadores de eficacia del microbioma.
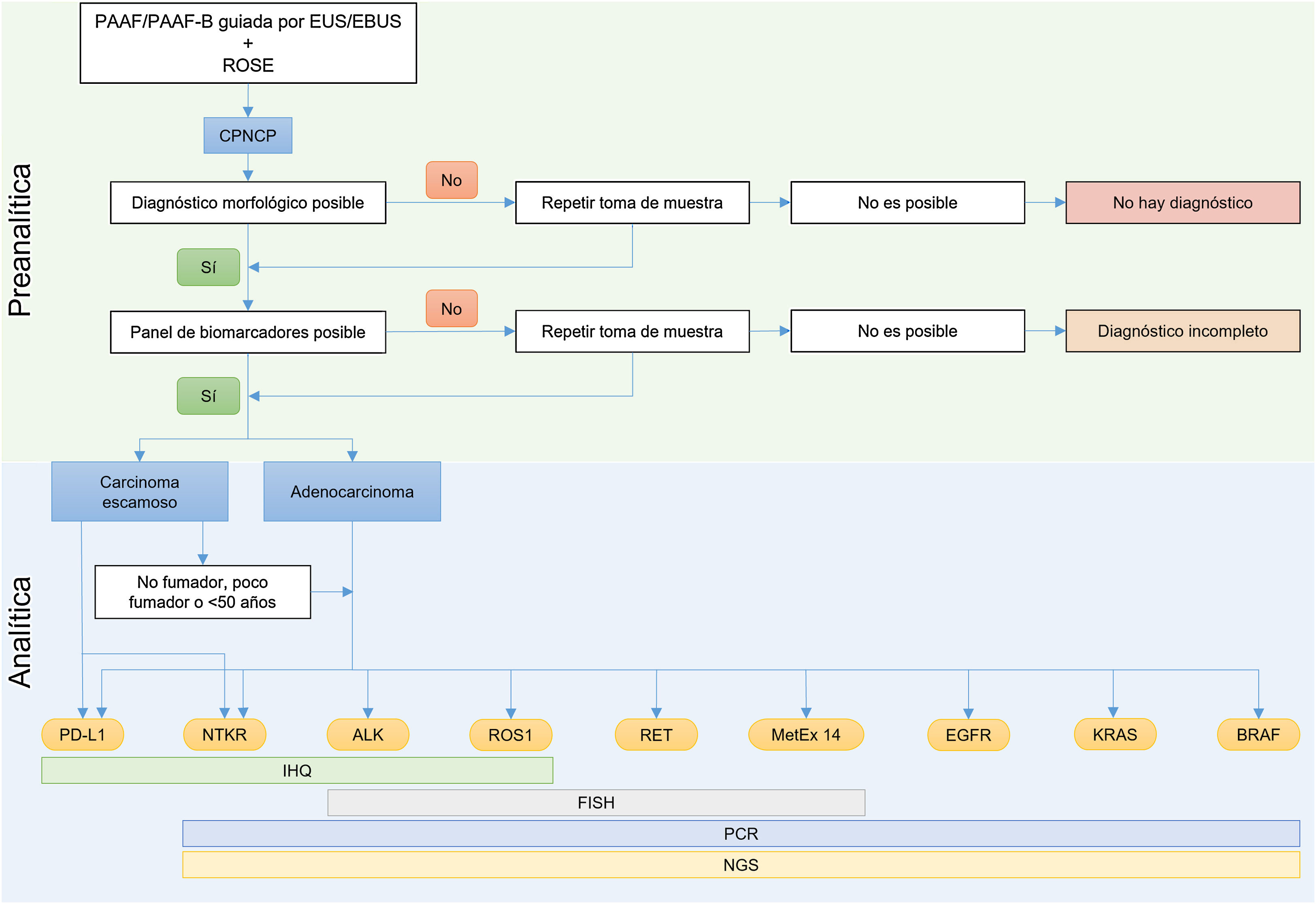
Priorizar el uso de muestras biológicas para lograr un diagnóstico precisoUn porcentaje alto de pacientes con CPNCP se diagnostica en estadios avanzados. Estos pacientes no son subsidiarios de cirugía, por lo que el diagnóstico se realiza mediante biopsias pequeñas y muestras citológicas. El desarrollo de técnicas de imagen que guían la punción de aspiración con aguja fina (PAAF) y la biopsia por aspiración con aguja fina (PAAF-B) permite la obtención de muestras en la cantidad y calidad necesarias para realizar un diagnóstico completo, tanto morfológico como de biomarcadores (fig. 1)5.
![Actualización del algoritmo diagnóstico con muestra pequeña en pacientes con CPNCP.[[[[FP]]]] ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; EBUS: endobronchial ultrasound; EGFR: epidermal growth factor receptor; EUS: endoscopic ultrasound; FISH: hibridación fluorescente in situ; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MetEx 14: mesenchymal epithelial transition factor exon 14; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PAAF: punción por aspiración con aguja fina; PAAF-B: biopsia por aspiración con aguja fina; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; ROS1: c-ros oncogene 1; RET: rearranged during transfection; ROSE: rapid on site evaluation.](https://static.elsevier.es/multimedia/16998855/0000005600000002/v2_202305072119/S1699885523000077/v2_202305072119/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNdV4B9vwZQjgKcZv3MP4CdsxvJsclGBCS9l8OAtxAyh+MFAB02CUXv6yohuVu/GmwQAXcDKi8D+S3tPmufPMWRlkKhJgXcgb9/qSCGafCZ4zDHweIy0vJhdZtCEDsYBl4M5I2HGWcACHGeWQY34PfRDi4pcGE+1EifMzF1YDCo4C8PghFD8GEt9x65aa8IwHnzsC2VBursojQ7yZ5X1lzb7CIE1+9Ne41qhabevOpawF+sG7xzp2m1L47kzHlSurs8= "Actualización del algoritmo diagnóstico con muestra pequeña en pacientes con CPNCP.[[[[FP]]]] ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; EBUS: endobronchial ultrasound; EGFR: epidermal growth factor receptor; EUS: endoscopic ultrasound; FISH: hibridación fluorescente in situ; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MetEx 14: mesenchymal epithelial transition factor exon 14; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PAAF: punción por aspiración con aguja fina; PAAF-B: biopsia por aspiración con aguja fina; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; ROS1: c-ros oncogene 1; RET: rearranged during transfection; ROSE: rapid on site evaluation.")
Actualización del algoritmo diagnóstico con muestra pequeña en pacientes con CPNCP.[[[[FP]]]] ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; EBUS: endobronchial ultrasound; EGFR: epidermal growth factor receptor; EUS: endoscopic ultrasound; FISH: hibridación fluorescente in situ; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MetEx 14: mesenchymal epithelial transition factor exon 14; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PAAF: punción por aspiración con aguja fina; PAAF-B: biopsia por aspiración con aguja fina; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; ROS1: c-ros oncogene 1; RET: rearranged during transfection; ROSE: rapid on site evaluation.
Las guías internacionales recomiendan, independientemente del tipo de muestra77: i) realizar un diagnóstico morfológico preciso (es decir, subtipificar el CPNCP); ii) el diagnóstico de CPNCP subtipo no especificado (CPNCP-NE) debe ser menor a 10% de los diagnósticos con muestra pequeña/citología; iii) hacer un uso juicioso de la IHQ/inmunocitoquímica (immunocytochemistry [ICC]); y iv) reservar muestra para estudio de biomarcadores.
En el caso de la punción PAAF/PAAF-B guiada por técnicas de imagen, se recomienda el uso de la evaluación rápida in situ (rapid on-site evaluation [ROSE]). Además de evaluar si una muestra es suficientemente adecuada para facilitar una aproximación diagnóstica, ROSE también permite controlar toda la fase preanalítica y la preparación in situ de la muestra para el análisis de los biomarcadores necesarios según la impresión diagnóstica preliminar5,78.
En el caso de las muestras citológicas, se recomienda manejar la muestra in situ para procesarla de forma adecuada mediante: i) extensiones con fijación en alcohol de 96° inmediata; ii) extensiones secadas al aire y teñidas con Giemsa®/Diff-Quik®; iii) bloque celular; iv) lavado de aguja en fijador de citología líquida (lo que proporciona buena conservación de ARN).
Cualquiera de estos tipos de muestra citológica es útil para la realización de biomarcadores mediante IHQ/ICC, FISH y técnicas basadas en PCR5,79–81.
La IHQ/ICC ofrece resultados excelentes, que son comparables a los obtenidos mediante biopsia en bloque celular y en extensiones previamente teñidas con Papanicolaou. Las extensiones citológicas no teñidas y teñidas con Giemsa®/Diff-Quik® y Papanicolaou son un sustrato excelente para FISH. Se analizan núcleos enteros, evitando los problemas relacionados con el corte en el material de parafina (es decir, los núcleos no se truncan al cortar los bloques de parafina con el micrótomo, no se pierde la tridimensionalidad). El ADN y ARN es de mejor calidad en muestras no fijadas en formol.
El estudio molecular supone menor problema habitualmente en las piezas quirúrgicas, ya que se cuenta con mayor cantidad de tejido. No por esto está exenta de dificultades, debiendo realizarse una adecuada fijación de las piezas quirúrgicas, no menos de 24-48 h. Deben evitarse áreas de necrosis y realizar las determinaciones sobre muestras que cuenten con, al menos, con 30% de celularidad tumoral viable. Además, debe realizarse un tallado adecuado de las piezas, según protocolos macroscópicos (por ejemplo, Libro blanco de anatomía patológica), incluyendo un número suficiente de secciones del tumor. Los tumores de tamaño no mayor a 3 cm deben incluirse en su totalidad. Un buen estudio histológico es el primer biomarcador, ya que determinará el subtipo histológico y orientará las determinaciones moleculares a realizar (superponible a lo indicado anteriormente para biopsia pequeña y citología). Algunos subtipos histológicos están asociados a distintas alteraciones moleculares, si bien, no debe considersrse excluyente para la deteminación de biomarcadores en cáncer de pulmón ninguna variable clínica ni histológica. Otro debate se plantea acerca de la deteminación molecular en tumores con diferentes subtipos histológicos (hecho muy frecuente en adenocarcinomas). En estos casos sería conveniente testar en la muestra correspondiente al subtipo más frecuente, añadiendo además la determinación en subtipos secundarios, sobre todo si presentan diferenciación mucinosa, en células claras, anillo de sello o presencia de psammomas.
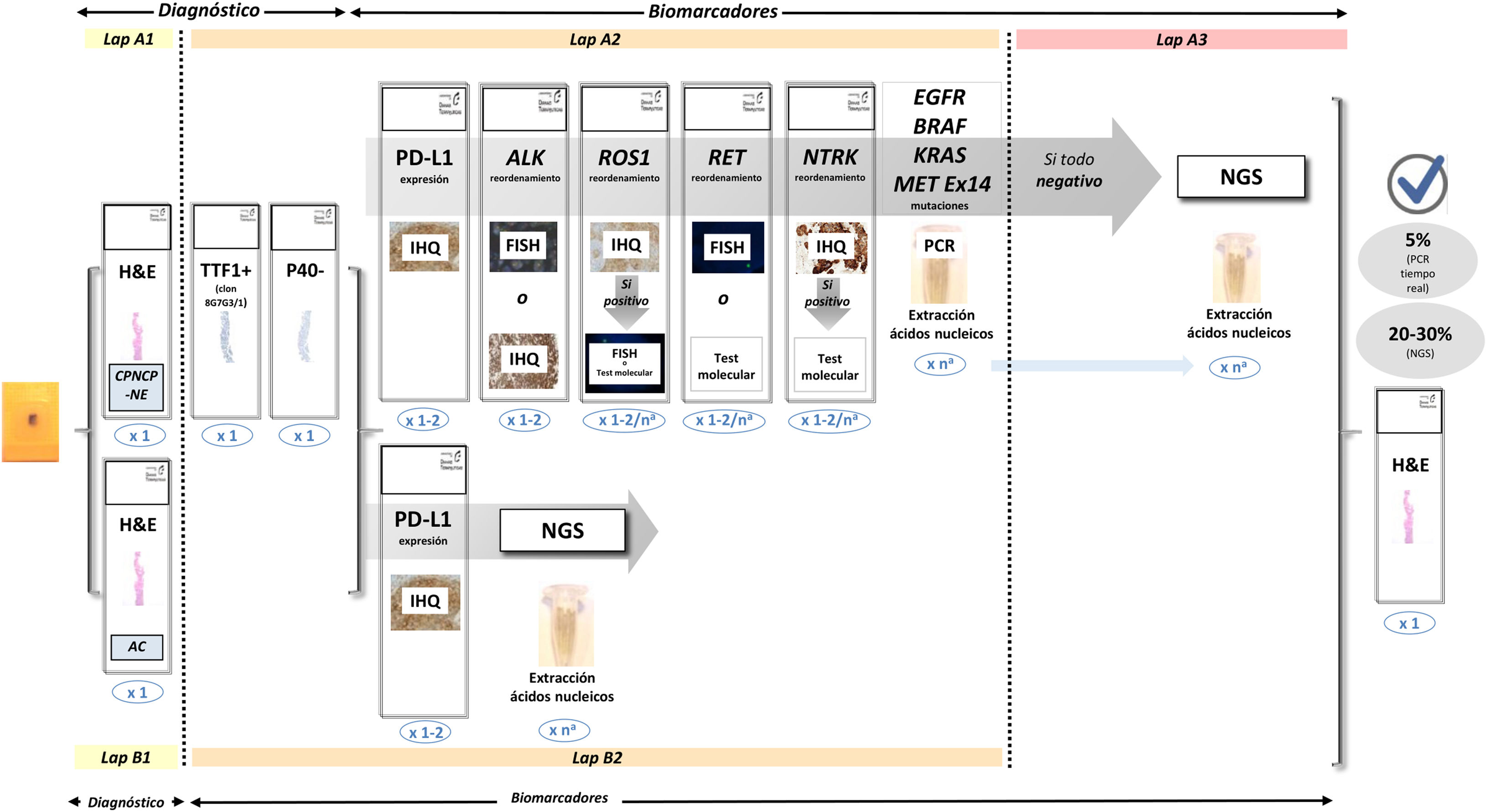
Para todos los métodos de análisis de biomarcadores citados anteriormente, en la figura 2 se expone una actualización del protocolo a seguir para analizar una muestra biológica de CPNCP.
![Actualización del protocolo para el análisis de múltiples biomarcadores en una muestra biológica de CPNCP.[[[[FP]]]]El número de secciones de cada prueba se muestra en azul. aLos requisitos para la extracción de ácido nucleico en pruebas moleculares individuales o paneles genéticos extendidos (NGS) son variables. Figura modificada de Conde E, et al. (confidencial, enviado). AC: adenocarcinoma; ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; CPNCP-NE: CPNCP subtipo no especificado; EGFR: epidermal growth factor receptor; FISH: hibridación fluorescente in situ; H&E: haematoxylin and eosin; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MET: mesenchymal epithelial transition factor; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; RET: rearranged during transfection; ROS1: c-ros oncogene 1.](https://static.elsevier.es/multimedia/16998855/0000005600000002/v2_202305072119/S1699885523000077/v2_202305072119/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNdV4B9vwZQjgKcZv3MP4CdsxvJsclGBCS9l8OAtxAyh+MFAB02CUXv6yohuVu/GmwQAXcDKi8D+S3tPmufPMWRlkKhJgXcgb9/qSCGafCZ4zDHweIy0vJhdZtCEDsYBl4M5I2HGWcACHGeWQY34PfRDi4pcGE+1EifMzF1YDCo4C8PghFD8GEt9x65aa8IwHnzsC2VBursojQ7yZ5X1lzb7CIE1+9Ne41qhabevOpawF+sG7xzp2m1L47kzHlSurs8= "Actualización del protocolo para el análisis de múltiples biomarcadores en una muestra biológica de CPNCP.[[[[FP]]]]El número de secciones de cada prueba se muestra en azul. aLos requisitos para la extracción de ácido nucleico en pruebas moleculares individuales o paneles genéticos extendidos (NGS) son variables. Figura modificada de Conde E, et al. (confidencial, enviado). AC: adenocarcinoma; ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; CPNCP-NE: CPNCP subtipo no especificado; EGFR: epidermal growth factor receptor; FISH: hibridación fluorescente in situ; H&E: haematoxylin and eosin; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MET: mesenchymal epithelial transition factor; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; RET: rearranged during transfection; ROS1: c-ros oncogene 1.")
Actualización del protocolo para el análisis de múltiples biomarcadores en una muestra biológica de CPNCP.[[[[FP]]]]El número de secciones de cada prueba se muestra en azul. aLos requisitos para la extracción de ácido nucleico en pruebas moleculares individuales o paneles genéticos extendidos (NGS) son variables. Figura modificada de Conde E, et al. (confidencial, enviado).
AC: adenocarcinoma; ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; CPNCP: carcinoma de pulmón de células no pequeñas; CPNCP-NE: CPNCP subtipo no especificado; EGFR: epidermal growth factor receptor; FISH: hibridación fluorescente in situ; H&E: haematoxylin and eosin; IHQ: inmunohistoquímica; KRAS: kirsten rat sarcoma virus; MET: mesenchymal epithelial transition factor; NGS: next-generation sequencing; NTRK: neurotrophic tyrosine receptor kinase; PCR: polymerase chain reaction; PD-L1: programmed death ligand-1; RET: rearranged during transfection; ROS1: c-ros oncogene 1.
En los últimos años se ha incrementado la necesidad de realizar pruebas multigénicas en pacientes con cáncer de pulmón, incluyendo alteraciones de factores oncogénicos, comutaciones o mecanismos de resistencia82. La NGS permite secuenciar genes largos y complejos y múltiples genes en una muestra del paciente, con el fin de identificar alteraciones de factores y dianas, minimizando el uso de tejidos en un corto periodo de tiempo, y su uso en la práctica clínica diaria.
La Sociedad Europea de Oncología Médica (ESMO) ha establecido recomendaciones sobre si la NGS multigénica tumoral puede utilizarse y cómo para perfilar los cánceres metastásicos siguiendo la clasificación de la Escala de Capacidad de actuación Clínica de Objetivos Moleculares (Scale for Clinical Actionability of Molecular Targets [ESCAT])83. La ESCAT es un marco que clasifica la correspondencia entre el fármaco y las alteraciones genómicas, según su capacidad de actuación a tres niveles84: i) desde la perspectiva de la salud pública; ii) desde la perspectiva de los centros académicos de investigación clínica; y iii) a nivel de cada paciente individual.
En lo que respecta al cáncer de pulmón, las recomendaciones generales para la práctica diaria consideran que una muestra de tumor o plasma de un paciente con NSCLC no escamoso avanzado se perfila mediante tecnología NGS, con el fin de detectar las alteraciones ESCAT de nivel I (el emparejamiento alteración-fármaco se asocia con un mejor resultado en los ensayos clínicos, listos para uso rutinario). Las pruebas deben incluir EGFR, ALK, ROS1, BRAF, RET, HER2, NTRK, KRAS y MET. Además, para los centros de investigación clínica, es muy recomendable realizar la secuenciación multigénica para acceder a fármacos innovadores y ensayos clínicos, a diferencia del uso en pacientes individuales, para los que se esperan pocos hallazgos clínicamente significativos con la NGS83.
La elección del tamaño del panel de NGS dependerá del tipo de alteraciones que se quieran estudiar, del tiempo de respuesta que se requiera y de los costes que se puedan esperar85,86. Se recomienda que cada servicio implemente el panel que mejor cubra sus necesidades y que se esté muy familiarizado con su cobertura para poder ampliar los estudios moleculares si los resultados iniciales son completamente negativos. En este sentido, para identificar fusiones tratables se recomienda el uso de paneles de ARN debido a que son más sensibles que los que usan exclusivamente ADN87.
Los ácidos nucleicos de partida se obtienen principalmente de muestras fijadas en formol e incluidas en parafina (englobando tanto las muestras tisulares como los bloques celulares procedentes de muestras citológicas) o de los ácidos nucleicos presentes en el plasma. En la primera etapa de preparación de la muestra, es fundamental la revisión exhaustiva de todo el material de cada paciente para la selección del bloque parafinado más adecuado, teniendo en cuenta las variables preanalíticas (hay que evitar la fijación insuficiente y todos los fijadores que no sean formol neutro tamponado a 10%) y el porcentaje de celularidad tumoral (punto de corte óptimo: igual o mayor a 30%)88.
Las dos aproximaciones metodológicas de NGS más implementadas en la práctica clínica son la amplificación en puente (Illumina®, San Diego, CA, EE. UU.) y la PCR en emulsión (Thermo Fisher Scientific®, Waltham, MA, EE. UU.), habiéndose descrito fortalezas y debilidades para cada una de ellas. Entre las ventajas de la amplificación por puente de Illumina® se destaca que permite identificar alteraciones «desconocidas» y trabajar con paneles de genes más grandes, mientras que la PCR en emulsión de Thermo Fisher Scientific® requiere menor cantidad de material de partida y emite resultados moleculares con tiempos de respuesta más cortos54,89.
Los hallazgos moleculares obtenidos se deben reflejar en el informe de resultados de la NGS, junto con las conclusiones relevantes en el contexto del tumor de cada paciente. Es muy importante la discusión de los resultados en un comité multidisciplinar, ya que existen cada vez más evidencias de que esta práctica mejora los resultados clínicos90.
El papel de la biopsia líquida en el CPNCPEl concepto de biopsia líquida define aquellas pruebas realizadas sobre una muestra de sangre periférica u otro fluido biológico con el objetivo de detectar en ellos células tumorales circulantes (CTC) o fragmentos de ácidos nucleicos procedentes de un tumor como ADN libre circulante (cfADN), ctADN, exosomas circulantes, ARN plaquetario y ARN circulante tumoral (ctARN), aislados en sangre (plasma) o en orina, líquidos pleurales, ascíticos, líquido cefalorraquídeo (LCR) y saliva. La biopsia líquida alcanza una especificidad elevada (de 96%) pero su sensibilidad baja hasta 66%91, por lo que un resultado negativo no es definitivo y precisa una confirmación sobre el tejido.
Por tanto, su papel es complementario a la biopsia y sus recomendaciones actuales se basan en dos contextos clínicos cuando el tejido sea limitado o insuficiente: a) la determinación de alteraciones moleculares que afectan a la sensibilidad al tratamiento, y b) la determinación de los mecanismos de resistencia tras progresión con un TKI21,92. Un tercer contexto sería la monitorización de la eficacia del tratamiento basada en carga de ctADN para enfermedad mínima residual (EMR), aproximación atractiva pero todavía no bien establecida desde el punto de vista técnico (es necesario establecer las unidades de cuantificación), pero donde los niveles fluctuantes de ADN circulante no tumoral pueden afectar los resultados92. Su uso en diagnóstico precoz presenta dificultades por la baja sensibilidad en enfermedad localizada. Básicamente, el mayor desarrollo se ha hecho en mutaciones de EGFR, pero actualmente se está incorporando en otras alteraciones moleculares, aunque la detección de reordenamientos génicos a partir de ARN circulante continúa siendo un reto técnico en vías de resolución21.
Algunos requerimientos técnicos de la biopsia líquida son la necesidad de volúmenes de sangre mayores de lo habitual (dos tubos de 10 mL). Es preferible utilizar plasma mejor que suero para la extracción de ácidos nucleicos. El tiempo máximo de espera hasta la extracción del plasma es de dos horas para tubos con ácido etilendiaminotetraacético (EDTA) y tres días para tubos con agentes preservadores especiales (Streck®). No se debe congelar la sangre antes de la extracción del plasma. La extracción del ADN debe realizarse con protocolos diseñados para ADN de pequeño tamaño y fragmentado92. También recordar que hasta 10% de mayores de 65 años presentan fenómenos de hematopoyesis clonal que podría ser malinterpretada como falsos positivos93.
Se recomienda el uso de técnicas con elevada sensibilidad, como la PCR digital. En el caso de la NGS hay una buena concordancia con los resultados en tejido, excepto para las variantes que se encuentran con una frecuencia alélica menor de 1%94, si bien existen herramientas como el identificador molecular único (unique molecular identifier [UMI]) para optimizar la detección. Dos plataformas comerciales de NGS (Guardant360® y FoundationOne Liquid CDx) tienen la aprobación de la FDA para el análisis de tumores sólidos incluyendo carcinomas de pulmón21.
En definitiva, la biopsia líquida se irá progresivamente incorporando al diagnóstico molecular, al seguimiento del tratamiento, a la detección de EMR y al diagnóstico precoz en cuanto los estudios prospectivos confirmen su utilidad clínica.
Principales requisitos para implementar un buen control de calidadLas pruebas moleculares se están convirtiendo en una herramienta de diagnóstico esencial y en una parte del tratamiento estándar en pacientes con cáncer. Tanto los laboratorios como los patólogos se enfrentan a nuevos retos para poder cumplir con este novedoso requisito en la atención al paciente. Los laboratorios de patología deben incorporar métodos fiables para garantizar una calidad y un procesamiento óptimos de las muestras a fin de reducir el riesgo de errores en las pruebas de biología molecular95. Además, los patólogos que ejercen deben ir más allá del diagnóstico y la clasificación para producir la información necesaria para guiar el tratamiento de manera precisa y oportuna96.
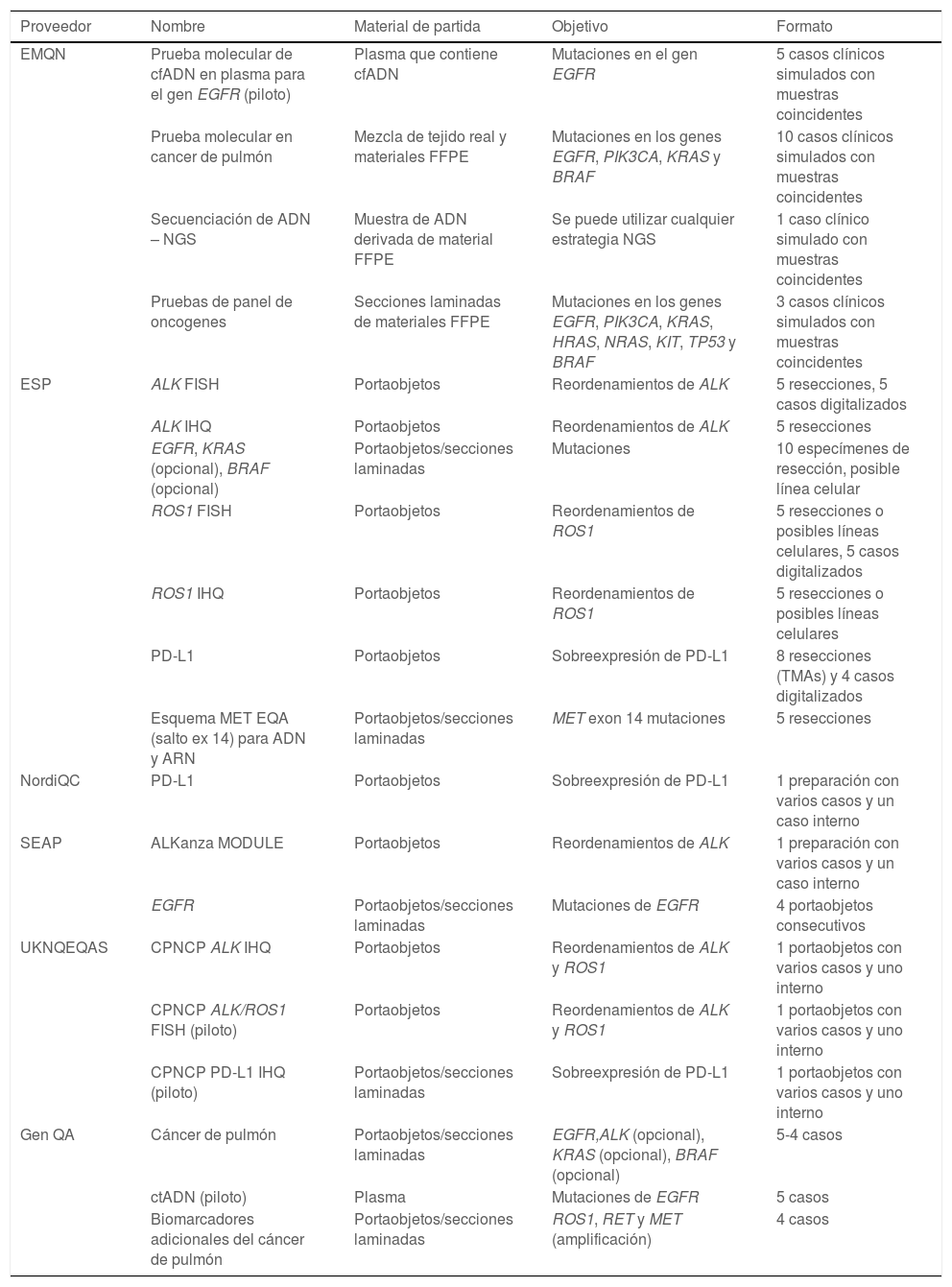
Los resultados de los biomarcadores predictivos a menudo determinan qué terapia (por ejemplo, quimioterapia, inmunoterapia o terapia dirigida) reciben los pacientes. Por lo tanto, los errores de laboratorio pueden derivar en decisiones de tratamiento incorrectas o subóptimas y, en consecuencia, dañar al paciente. Para garantizar pruebas de alta calidad, los laboratorios deben contar con un sistema de garantía de calidad y cumplir con los estándares internacionales relevantes de organizaciones certificadas como la Organización Internacional para la Estandarización (International Organization for Standardization [ISO]), el Colegio de Patólogos Estadounidenses (College of American Pathologists [CAP]) o el programa de las Enmiendas para el Mejoramiento de Laboratorios Clínicos (Clinical Laboratory Improvement Amendments [CLIA]) (tabla 3)96–100.
Ejemplos de esquemas europeos de garantía de calidad
| Proveedor | Nombre | Material de partida | Objetivo | Formato |
|---|---|---|---|---|
| EMQN | Prueba molecular de cfADN en plasma para el gen EGFR (piloto) | Plasma que contiene cfADN | Mutaciones en el gen EGFR | 5 casos clínicos simulados con muestras coincidentes |
| Prueba molecular en cancer de pulmón | Mezcla de tejido real y materiales FFPE | Mutaciones en los genes EGFR, PIK3CA, KRAS y BRAF | 10 casos clínicos simulados con muestras coincidentes | |
| Secuenciación de ADN – NGS | Muestra de ADN derivada de material FFPE | Se puede utilizar cualquier estrategia NGS | 1 caso clínico simulado con muestras coincidentes | |
| Pruebas de panel de oncogenes | Secciones laminadas de materiales FFPE | Mutaciones en los genes EGFR, PIK3CA, KRAS, HRAS, NRAS, KIT, TP53 y BRAF | 3 casos clínicos simulados con muestras coincidentes | |
| ESP | ALK FISH | Portaobjetos | Reordenamientos de ALK | 5 resecciones, 5 casos digitalizados |
| ALK IHQ | Portaobjetos | Reordenamientos de ALK | 5 resecciones | |
| EGFR, KRAS (opcional), BRAF (opcional) | Portaobjetos/secciones laminadas | Mutaciones | 10 especímenes de resección, posible línea celular | |
| ROS1 FISH | Portaobjetos | Reordenamientos de ROS1 | 5 resecciones o posibles líneas celulares, 5 casos digitalizados | |
| ROS1 IHQ | Portaobjetos | Reordenamientos de ROS1 | 5 resecciones o posibles líneas celulares | |
| PD-L1 | Portaobjetos | Sobreexpresión de PD-L1 | 8 resecciones (TMAs) y 4 casos digitalizados | |
| Esquema MET EQA (salto ex 14) para ADN y ARN | Portaobjetos/secciones laminadas | MET exon 14 mutaciones | 5 resecciones | |
| NordiQC | PD-L1 | Portaobjetos | Sobreexpresión de PD-L1 | 1 preparación con varios casos y un caso interno |
| SEAP | ALKanza MODULE | Portaobjetos | Reordenamientos de ALK | 1 preparación con varios casos y un caso interno |
| EGFR | Portaobjetos/secciones laminadas | Mutaciones de EGFR | 4 portaobjetos consecutivos | |
| UKNQEQAS | CPNCP ALK IHQ | Portaobjetos | Reordenamientos de ALK y ROS1 | 1 portaobjetos con varios casos y uno interno |
| CPNCP ALK/ROS1 FISH (piloto) | Portaobjetos | Reordenamientos de ALK y ROS1 | 1 portaobjetos con varios casos y uno interno | |
| CPNCP PD-L1 IHQ (piloto) | Portaobjetos/secciones laminadas | Sobreexpresión de PD-L1 | 1 portaobjetos con varios casos y uno interno | |
| Gen QA | Cáncer de pulmón | Portaobjetos/secciones laminadas | EGFR,ALK (opcional), KRAS (opcional), BRAF (opcional) | 5-4 casos |
| ctADN (piloto) | Plasma | Mutaciones de EGFR | 5 casos | |
| Biomarcadores adicionales del cáncer de pulmón | Portaobjetos/secciones laminadas | ROS1, RET y MET (amplificación) | 4 casos |
ALK: anaplastic lymphoma kinase; BRAF: B-Raf proto-oncogene; cfADN: ADN libre circulante; CPNCP: carcinoma de pulmón no célula pequeña; ctADN: ADN tumoral circulante; EGFR: epidermal growth factor receptor; FFPE: formalin-fixed paraffin-embedded; FISH: hibridación fluorescente in situ; HRAS: Harvey Rat sarcoma virus; IHQ: inmunohistoquímica; KIT: proto-oncogene receptor tyrosine kinase; KRAS: kirsten rat sarcoma virus; MET: mesenchymal epithelial transition factor; NGS: next-generation sequencing; NRAS: neuroblastoma ras viral oncogene homolog; PD-L1: programmed death ligand-1; PIK3CA: phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha; RET: rearranged during transfection; ROS1: c-ros oncogene 1; TMAs: tissue microarrays; TP53: tumour protein 53.
El progreso de la medicina personalizada se ve limitado, en parte, por la falta de documentación europea e internacional normalizada y la insuficiencia de guías para los flujos de trabajo preanalíticos. El proceso preanalítico ha sido examinado recientemente con cierto detalle por el proyecto SPIDIA® (http://www.spidia.eu). Los siguientes datos se consideraron necesarios para emitir un informe conforme a las guías de buenas prácticas (tabla 4)96: i) identificación del paciente: el paciente debe estar correctamente identificado –los laboratorios exigen un mínimo de dos identificadores únicos del paciente más un identificador único de la muestra en el formulario de solicitud y en el informe–; ii) estilo y contenido de los informes: los informes largos rara vez se leen en su totalidad, y la longitud importa; informes de una página o, mejor aún, de una sola cara, siempre que sean legibles. Debe incluirse una presentación clara de los resultados, la(s) prueba(s) realizada(s) y cualquier limitación de las pruebas (por ejemplo, ¿se analizaron todas las mutaciones posibles o solo una selección de las más comunes?); iii) interpretación: debe describirse el resultado de la prueba y proporcionársele una interpretación adecuada, sobre todo cuando se trate de una decisión terapéutica; iv) informes integrados: la necesidad de integrar los resultados de los pacientes está ampliamente reconocida. A medida que se generalice el uso de paneles genéticos, los resultados de las distintas pruebas genéticas deberán fusionarse en un único informe. Además, los resultados de varias especialidades patológicas sobre pacientes individuales deben integrarse en el mismo informe.
Propuesta de informe de resultados patológicos
| Identificación del paciente y del médico que solicitó la prueba (o, en su defecto, de la persona autorizada) |
| Diagnóstico patológico |
| Tipo de muestra presentada |
| Tratamiento previo (Sí/No) |
| Momento de la biopsia (inicial/recaída/progresión) |
| Fecha de recogida de la muestra |
| El código externo en el caso de los centros de referencia |
| El medio en el que se recibió la muestra (fresca, congelada, embebida en parafina, etc.) |
| El origen anatómico de la muestra |
| La fecha del envío, la fecha de recepción de las muestras y la fecha de emisión de los resultados |
| El método de prueba de biomarcadores utilizado, especificando las mutaciones detectables u otras anomalías. En el caso de los kits comerciales, deberá indicarse el nombre comercial, el número de lote y si se trata de un producto autorizado para «diagnóstico in vitro» |
| Nombre de la plataforma utilizada y fecha de caducidad del producto |
| La calidad de la muestra, especificando el porcentaje de células cancerosas y si la muestra se enriqueció mediante microdisección o macrodisección, así como la concentración y pureza del ADN |
| Comentarios sobre el carácter adecuado o inadecuado de la muestra |
| El resultado de la prueba, que define el tipo de anomalía molecular detectada o la ausencia de anomalías moleculares |
| Identifiación del profesional responsable de la prueba (en todas las fases) |
| Identificación del supervisor del laboratorio (opcional) |
| Cualquier información adicional o comentario de interés para el médico que solicitó la muestra |
| Acreditación o participación en programas de calidad |
Los servicios de patología quirúrgica deberían obtener una acreditación en material de garantía de calidad. Todos los laboratorios que prestan servicios de patología molecular deberían tener una acreditación de laboratorio según la normativa ISO 15189 o su equivalente nacional. La acreditación proporciona a los pacientes, al personal, a los usuarios de los servicios y a los comisionados pruebas de la competencia del laboratorio.
ConclusionesEn la actualidad, en pacientes con CPNCP es preciso definir una estrategia diagnóstica genómica clara que permita establecer indicaciones terapéuticas óptimas en cada uno.
Con este objetivo, en el nuevo consenso de la SEOM y la SEAP se proponen las siguientes recomendaciones: i) es mandatorio determinar las mutaciones EGFR, BRAF, KRAS y MET, las traslocaciones ALK, ROS1, RET y NTRK, y la expresión de PD-L1 en CPNCP; ii) otras determinaciones emergentes como la mutación HER2 y los biomarcadores inmunes como TMB, MSI, STK11 y KEAP1 son recomendables, especialmente si se dispone de NGS; iii) estas determinaciones moleculares se pueden realizar en cualquier estadio de CPNCP o pacientes clínicamente seleccionados y, de forma progresiva, se pueden establecer nuevas indicaciones terapéuticas que precisen de esta información; iv) la disponibilidad de NGS facilita en gran medida el diagnóstico molecular de forma precisa y eficaz, por lo que su uso debería generalizarse hoy en día; v) la biopsia líquida tiene un papel cada vez mayor en el diagnóstico molecular, especialmente si el tejido es limitado, pero también su papel en el seguimiento del tratamiento es prometedor, tanto en la detección de EMR y diagnóstico precoz; vi) es fundamental analizar de forma óptima la muestra tumoral, realizando una correcta priorización de las determinaciones moleculares que se van a realizar, así como un buen control de calidad en todo el proceso; vii) se necesita que exista una colaboración multidisciplinar adecuada entre los diferentes profesionales implicados para conseguir la máxima calidad en el proceso diagnóstico y en la determinación de la mejor oportunidad terapéutica para cada paciente con CPNCP en cualquier estadio de su enfermedad.
Contribución de los autoresTodos los autores contribuyeron a la elaboración y el diseño del proyecto. Todos los autores participaron en la redacción del primer borrador del manuscrito, comentaron los siguientes borradores y aprobaron la versión final del manuscrito.
FinanciaciónSEOM y SEAP agradecen el apoyo financiero de este proyecto en forma de colaboración sin restricciones en la logística por parte de AstraZeneca y Roche.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen la asistencia editorial de Beatriz Gil-Alberdi de HealthCo Trials (Madrid, España) en el desarrollo de este manuscrito.
Traducido de: Clinical and Translational Oncology. Isla D, Lozano MD, Paz-Ares L, Salas C, de Castro J, Conde E, Felip E, Gómez-Román J, Garrido P, Enguita AB. New update to the guidelines on testing predictive biomarkers in non-small-cell lung cancer: a National Consensus of the Spanish Society of Pathology and the Spanish Society of Medical Oncology. Clin Transl Oncol. 2022 Dec 26. doi: 10.1007/s12094-022-03046-9.

