




Primary hepatic liposarcoma is an extremely rare malignant tumour derived from adipocytes and is part of the group of mesenchymal tumours. We present the case of a 43-year-old Hispanic male patient with a pleomorphic hepatic liposarcoma and absence of MDM2 gene amplification. Two years and six months after surgery, the patient is asymptomatic. The present case is the first report of this entity with positive immunohistochemical testing for p16, p53, S100, vimentin and absence of MDM2 gene amplification.
El liposarcoma hepático primario es un tumor maligno extremadamente raro, derivado de adipocitos, y forma parte del grupo de tumores mesenquimales. Presentamos el caso de un paciente masculino de 43 años con diagnóstico de liposarcoma hepático pleomorfo con ausencia de amplificación del gen MDM2. Dos años y 6 meses después de la cirugía el paciente se encuentra asintomático. El presente caso es el primer informe de esta entidad con estudio inmunohistoquímico positivo para p16, p53, S100, vimentina y ausencia de amplificación del gen MDM2.
Liposarcoma is one of the most frequent malignant tumours of the soft tissue sarcoma (STS) group; it is derived from adipocytes and accounts for 15–20% of all STS. The World Health Organization (WHO) groups liposarcoma within the malignant adipocytic tumours and further subclassifies it according to its histopathological characteristics into five groups: well-differentiated, dedifferentiated, myxoid, myxoid–pleomorphic and pleomorphic; the last one is the least frequent.1–3 Liposarcoma can metastasize in up to 30% of cases to the liver, lung, brain, and bone; however, there are no epidemiological data in the literature on its primary hepatic form. Weitz et al. mention that the prevalence of primary hepatic sarcomas is 1% despite the fact that, in their prospective study of 30 cases of primary hepatic sarcomas, none of the liposarcomas were included.4 Pleomorphic liposarcoma is considered a high-grade sarcoma, which mostly affects patients between 54 and 70 years of age; its diagnosis tends to be an unexpected finding in view of its most frequent presentation as an asymptomatic mass. The most common anatomical site is the lower extremities, followed by the trunk, retroperitoneum, and spermatic cord.5 Due to the extreme rarity of this tumour with primary hepatic localization and pleomorphic subtype, there is insufficient medical literature; consequently, diagnosis remains a challenge due to its histological and imaging similarity to other liposarcoma subtypes and the need to rule out other more common liver tumours. To the best of our knowledge, there are only two case reports of this entity.6,7 We present a case in an asymptomatic patient with primary hepatic pleomorphic liposarcoma, with expression of p16, p53 and non-amplified MDM2.
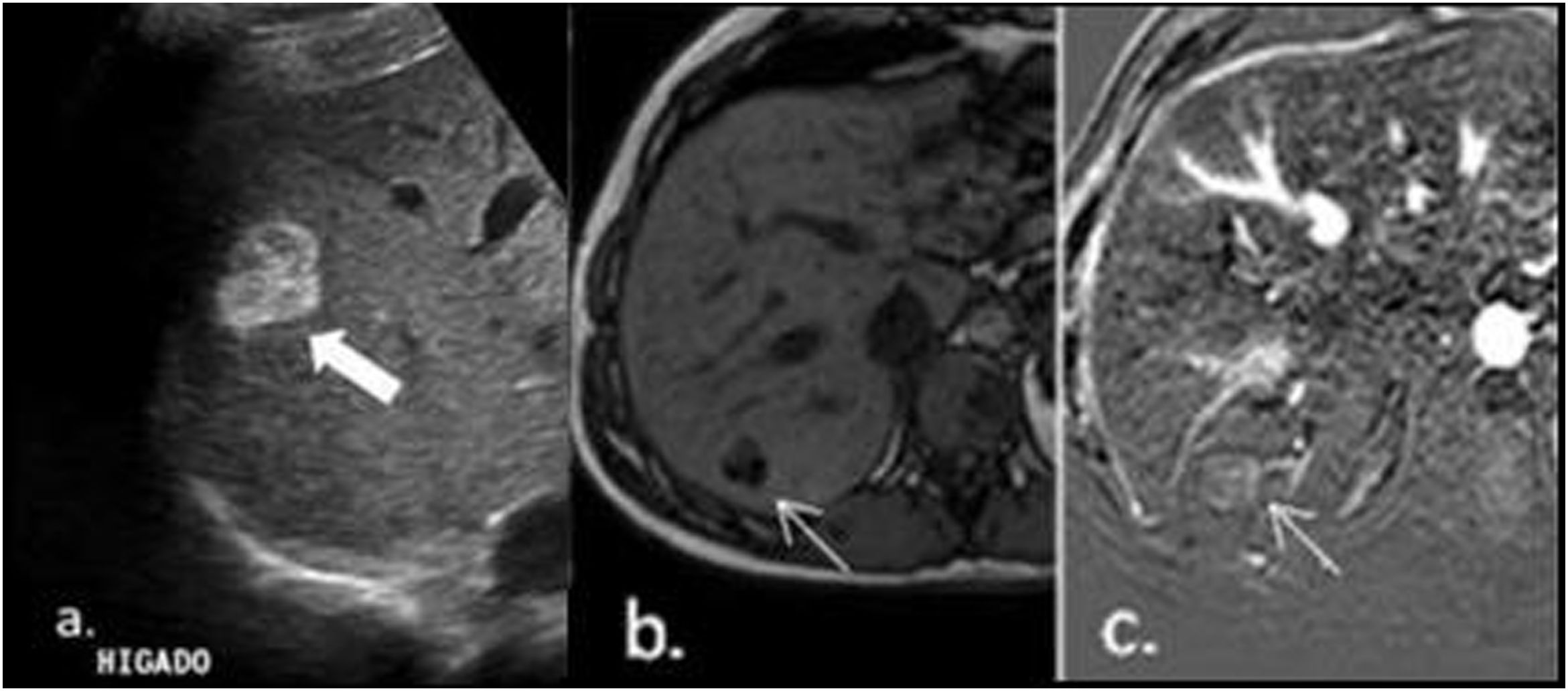
Case reportA 43-year-old Hispanic male patient with a history of atrial fibrillation, dyslipidaemia, and hepatic steatosis; an abdominal ultrasound was performed in order to comply with his occupational exams. Ultrasound reported a hyperechogenic indeterminate solid lesion in segment VII measuring 26mm×22mm (Fig. 1a). For a better characterization of this lesion, simple and contrast MRI of the abdomen was performed, which showed the nodule as hypervascular with intralesional fat (Fig. 1b and c). These findings in the context of a non-cirrhotic liver were interpreted as an adenoma.
. For a better characterization of this lesion, simple and contrast MRI of the abdomen was performed, which showed the nodule as hypervascular with intralesional fat (b and c). These findings in the context of a non-cirrhotic liver were interpreted as an adenoma.")
Ultrasound reported a hyperechogenic indeterminate solid lesion in segment VII measuring 26mm×22mm (a). For a better characterization of this lesion, simple and contrast MRI of the abdomen was performed, which showed the nodule as hypervascular with intralesional fat (b and c). These findings in the context of a non-cirrhotic liver were interpreted as an adenoma.
The absence of tumour findings in the retroperitoneum is noteworthy. Physical examination was normal; the coproparasitological test was positive for Blastocystis hominis, and negative for faecal occult blood and Giardia lamblia. Synchronous gastrointestinal tumour was ruled out by upper and lower endoscopy. Given the imaging findings and the risk of malignancy of hepatic adenoma in males, the oncology board (tumour board) decided to perform a partial hepatectomy of segment VII. The histopathological study reported pleomorphic liposarcoma. Its primary hepatic origin was proved due to the negative results of the complementary tests. Two years and six months after surgery, the patient is asymptomatic; control imaging studies of skull, chest, abdomen, and pelvis showed no recurrence.
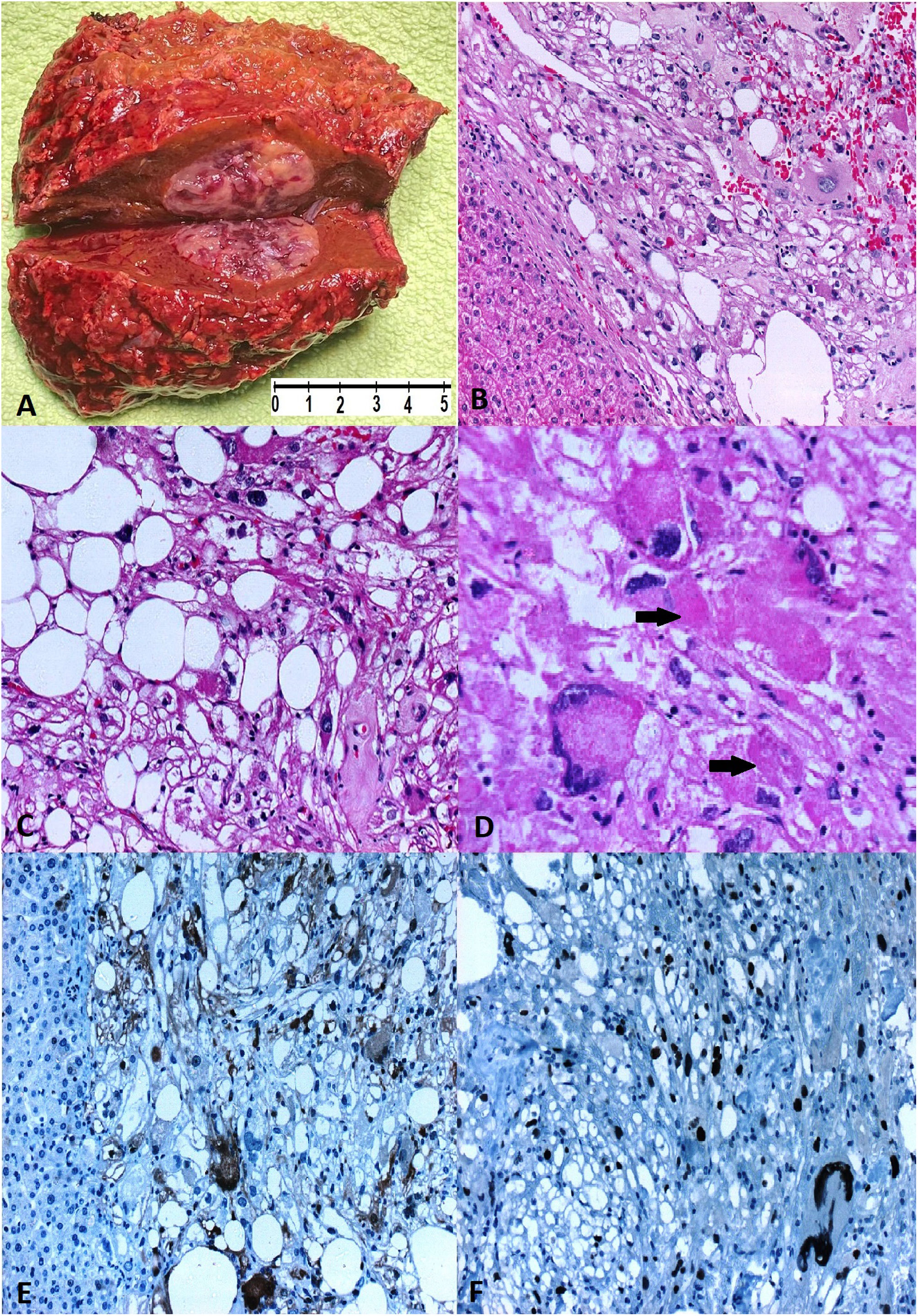
A segment of the right lobe of the liver, weighing 62.5g and measuring 7cm×6cm×3cm, was received. On cutting, a well-defined lesion of 2cm×2cm×1.5cm, friable, yellowish, with violaceous areas, was observed (Fig. 2A). Microscopically, the hepatic parenchyma was replaced by sarcomatous proliferation of predominantly spindle or giant epithelioid cells (Fig. 2B). It included a variable number of lipoblasts with hyperchromatic nuclei with marked pleomorphism and bordered by lipid cytoplasmic vacuoles (Fig. 2C). Frequent cytoplasmic hyaline structures (thanatosomes) were also observed (Fig. 2D). The mitotic index was 4 in 10 HPF, tumour necrosis of 30%, Ki-67 less than 10%, histological grade G2 according to (FNCLCC) and free surgical resection margins. Immunohistochemical staining showed negativity for CK-AE, Glypican-3, AFP and DOG1, and positivity for vimentin, p16 (no showed), S100 and p53 (Fig. 2E and F). In addition, absence of MDM2 gene amplification and microsatellite instability (MSI) were reported. The confirmed histopathological diagnosis was pleomorphic hepatic liposarcoma, TNM: pT1, pNx.
![Hepatic pleomorphic liposarcoma. (A) A white-yellow, relatively firm (2.2cm×2cm×1.5cm) nodular tumour on a hepatic segment. (B) A well-circumscribed, non-encapsulated tumour with infiltrative borders [HE 10×]. (C) Classic lipoblasts with scalloped nuclei or signet ring lipoblasts [HE 40×]. (D) Pleomorphic lipoblasts in a high-grade background with varying numbers of pleomorphic and frequently bizarre multinucleated tumour cells with intra-cytoplasmic “thanatosomes” (black arrows) [HE 40×]. (E) Diffuse cytoplasmic and nuclear S100 expression in tumour cells [20×]. (F) Diffuse p53 nuclear expression in tumour cells [40×].](https://static.elsevier.es/multimedia/16998855/0000005700000002/v1_202404090738/S1699885523000703/v1_202404090738/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNdV4B9vwZQjgKcZv3MP4CdslRkan0N7fAbLzbm9Mtgx77pSo3UI1o4hSaejELsvZkukMzaVZZZq7B57iwv71izaz9NDHHwXz5hRTTDlrfyYFiGbiuFShLLYFXfm6YU+aArZd9GYDgoV6vk/auG6nywUnhlz6KUUYSZotdRANUxD37LIKqTdTpo0z8PfnDjsnUjFwfTY3Bka8p2Jz8VGP2a4CnkbcKla71w2q2J3KZJqPXpFVN8Ucm4BLoKm2Rv/dMM= "Hepatic pleomorphic liposarcoma. (A) A white-yellow, relatively firm (2.2cm×2cm×1.5cm) nodular tumour on a hepatic segment. (B) A well-circumscribed, non-encapsulated tumour with infiltrative borders [HE 10×]. (C) Classic lipoblasts with scalloped nuclei or signet ring lipoblasts [HE 40×]. (D) Pleomorphic lipoblasts in a high-grade background with varying numbers of pleomorphic and frequently bizarre multinucleated tumour cells with intra-cytoplasmic “thanatosomes” (black arrows) [HE 40×]. (E) Diffuse cytoplasmic and nuclear S100 expression in tumour cells [20×]. (F) Diffuse p53 nuclear expression in tumour cells [40×].")
Hepatic pleomorphic liposarcoma. (A) A white-yellow, relatively firm (2.2cm×2cm×1.5cm) nodular tumour on a hepatic segment. (B) A well-circumscribed, non-encapsulated tumour with infiltrative borders [HE 10×]. (C) Classic lipoblasts with scalloped nuclei or signet ring lipoblasts [HE 40×]. (D) Pleomorphic lipoblasts in a high-grade background with varying numbers of pleomorphic and frequently bizarre multinucleated tumour cells with intra-cytoplasmic “thanatosomes” (black arrows) [HE 40×]. (E) Diffuse cytoplasmic and nuclear S100 expression in tumour cells [20×]. (F) Diffuse p53 nuclear expression in tumour cells [40×].
We performed immunohistochemical staining on 4μm formalin-fixed and paraffin-embedded (FFPE) tissue sections using the VENTANA Benchmark system (Roche, Tucson, AZ) according to standardized laboratory procedures. The following antibodies were used during the diagnostic study: CK-AE, Glypican-3, AFP, DOG1, vimentin, S100, p16, p53. For diagnostic and theranostic purposes, the amplification of the MDM2 gene was analyzed by in situ chromogenic hybridization (CISH), the result of which was negative. Detection of MSI Idylla™ test was performed.
DiscussionPleomorphic liposarcoma is the least frequent histopathological subtype of the group of soft tissue liposarcomas; it accounts for only 5% of these neoplasms, which are defined by adipocytic differentiation.8–12 Due to the scarce literature on the matter, the diagnosis of this condition remains a challenge because microscopic and imaging findings may mimic other sarcoma and liposarcoma subtypes. Its most frequent clinical manifestation is an asymptomatic mass that grows rapidly, while in some patients it causes pain or symptoms associated with compression of anatomical structures. Its molecular profile is complex and heterogeneous.3,5,8 Historically, the diagnosis of liposarcoma was based strictly on cell morphology due to the lack of diagnostic techniques to differentiate histological subtypes. However, reliance on morphology alone can lead to misinterpretation by pathologists and overlap with other liposarcomas and high-grade sarcomas in rare cases.10 For this reason, the main differential diagnosis of pleomorphic liposarcoma (PLS) is dedifferentiated liposarcoma. PLS is defined by the presence of pleomorphic, univacuolated or intra-cytoplasmic multivacuolated lipoblasts. Multinucleated and spindle giant cells, scalloped nuclei, clear cytoplasmatic vacuoles and nonlipoblastic pleomorphic sarcomatous cells are frequent, but not specific.10,13 Necrosis is evident in more than 50% of cases.3
Currently, immunohistochemistry, hybridization and molecular techniques have improved the ability to differentiate between the various subtypes of liposarcoma, high-grade sarcomas and benign mesenchymal tumours. In the case of pleomorphic liposarcoma, immunohistochemical staining shows positivity for S100 in lipoblasts and in certain cases, primarily for SMA, CD34, HMGA2, desmin, pankeratin, and EMA; and negativity for MDM2 and CDK4.14 Initially, the sarcomatous cell morphology with adipose component forced to consider a liposarcoma; therefore, a hybridization study (CISH) of the MDM2 gene was performed, which was negative. This result ruled out a dedifferentiated liposarcoma, in agreement with multiple case series that report the diagnostic and exclusionary usefulness of the amplification of this gene when comparing the pleomorphic subtype to the dedifferentiated one.9 Positive p16 and p53 have been described as diagnostic tools in other liposarcoma subtypes such as differentiated and dedifferentiated liposarcoma and have been poorly studied in the pleomorphic subtype due to their rarity. Ghadimi et al. described in their case series of 155 pleomorphic liposarcomas the p53 mutation in up to 55% of cases and p16 expression in 100% of cases.15 Although no case was primary hepatic, these results are similar to those of our patient, supporting the final diagnosis. The presence of sarcomatous cells in the liver parenchyma with intra- and extra-cytoplasmic hyaline bodies led us to consider the possibility of undifferentiated embryonal sarcoma. We ruled out this diagnosis on the basis of clinical, radiological, histopathological, immunohistochemical, and molecular evidence in favour of liposarcoma. Finally, Tachibana et al. reported the presence of cytoplasmic hyaline bodies in several entities, including a dedifferentiated pleomorphic sarcoma, reminding us that this histopathological finding is not exclusive to undifferentiated embryonal sarcoma.13,15
A contrast-enhanced computed tomography scan was requested to evaluate the lesion in more detail. This showed a hypervascular nodule with significant capsular contrast uptake. Given these findings, the lesion was interpreted as hepatocellular in origin; two possible diagnoses were considered, adenoma or focal nodular hyperplasia. In order to identify the cause of the lesion and define the diagnosis, hepatobiliary magnetic resonance imaging was performed with gadoxetic acid as a contrast agent. In-phase T1-weighted axial slice confirmed a hepatic nodule with foci of hyperintensity, followed by signal decay in the T1 opposed-phase sequence; these findings are consistent with a high-fat lesion. During the arterial phase, the lesion demonstrated significant signal uptake relative to liver parenchyma. During the portal phase there is washout, while in the apparent diffusion coefficient the lesion showed a low signal, characteristic of high cellularity. The fat-saturated image obtained 20min after contrast injection in the transverse T1-weighted hepatobiliary phase showed a lesion with lower signal intensity compared to the surrounding liver tissue; it is a typical finding that demonstrates the absence of functional hepatocytes.
It is essential to differentiate between focal nodular hyperplasia and adenoma for its treatment. Since the contrast cannot be excreted due to the absence of biliary canaliculi during the hepatobiliary phase, it leaves us with a hypervascular lesion without contrast retention, which enables us to rule out with high sensitivity and specificity focal nodular hyperplasia.10 Thus, in a 43-year-old male patient, with no history of anabolic steroids or other hormonal treatments, absence of chronic liver diseases, negative serology for hepatitis B and C virus, together with the imaging findings, led us to the preoperative radiological diagnosis of adenoma.
ConclusionPrimary pleomorphic hepatic liposarcoma is an extremely rare hepatic mesenchymal tumour. Because there are few reports in the medical literature, its clinical suspicion is nonexistent; this causes many cases to go unnoticed. Moreover, its clinical course is insidious and even the histopathological diagnosis can be confusing due to overlap with other histological subtypes. Thanks to the lessons learned from our case, we highlight the importance of multidisciplinary teams to establish the appropriate diagnosis and treatment. We recommend radiologists to suspect this diagnosis when faced with a hypervascular hepatic lesion, with a high-fat component, without contrast retention during the hepatobiliary phase, particularly in cases where clinically there is no suspicion of hepatocellular carcinoma. We also recommend that pathologists use hybridization studies to determine MDM2 gene amplification in order to confirm the pleomorphic subtype. Among primary hepatic sarcomas, the presence of intra- and extra-cytoplasmic hyaline structures is common in embryonal sarcoma and pleomorphic liposarcoma. p53 and p16 markers may be useful for this exceptional diagnosis; however, more cases like ours need to be reported so that, in the future, this poorly known condition can be taken into account in the differential diagnosis of hepatic mesenchymal lesions.
Ethical approvalThe patient agreed that his stored material was enclosed and that his clinical data were anonymously used.
Authors’ contributionsNelson Montalvo: Conceptualization, methodology, supervision, validation, writing – reviewing and editing. Ligia Redrobán: Supervision, validation, writing – reviewing and editing. Jorge Lara-Endara: Data curation, investigation and writing – original draft preparation. Christian Armijos: Resources and visualization. Franz Serpa: Resources and visualization. Javier Rodríguez-Suárez: Resources. All authors have approved the final version.
FundingNone declared.
Conflicts of interestThe authors declare that they have no conflicts of interest.

