


Descripción del caso clínico
Mujer de 43 años de edad, diagnosticada en 1999 de artritis reumatoide (AR) seropositiva erosiva, que ha seguido tratamiento con aurotiomalato oral, que se cambió en marzo de 2000 por metotrexato oral (dosis máxima de 12,5 mg/semana) y corticoides (prednisona 10 mg/día), terapia mantenida hasta el momento actual, con un regular control de la actividad articular. En marzo de 2003 se planteó iniciar tratamiento con infliximab para controlar la actividad de su enfermedad, pero la paciente lo rechazó. Tres meses después, la paciente acude a la consulta por aparición de una tumoración dolorosa en la región pretibial izquierda, de crecimiento progresivo en el último mes, sin referir otros datos clínicos acompañantes.
La exploración física puso de manifiesto una tumoración de consistencia blanda, dolorosa a la palpación y adherida a planos profundos, en la región proximal externa de la tibia izquierda. No se objetivaron adenopatías periféricas ni organomegalias. Presentaba sinovitis en ambos carpos y en las articulaciones metacarpofalángicas.
En las pruebas de laboratorio, el hemograma, la bioquímica, el proteinograma, las inmunoglobulinas y el sistemático de orina fueron normales. Únicamente destacaba una VSG de 32 mm/h y una PCR de 10,1 mg/l, ya objetivadas en revisiones previas.
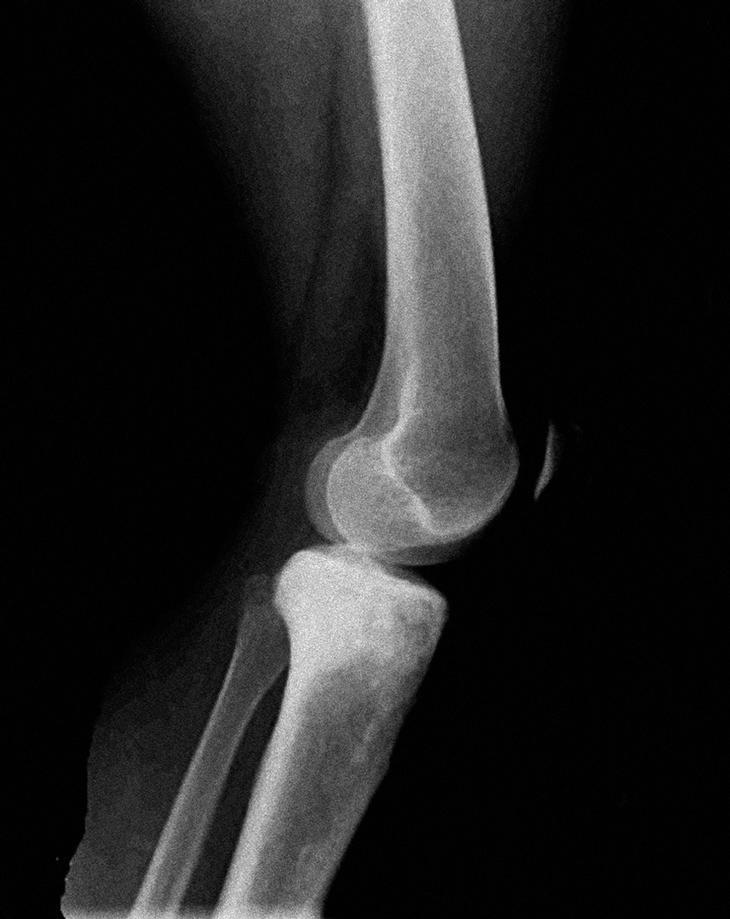
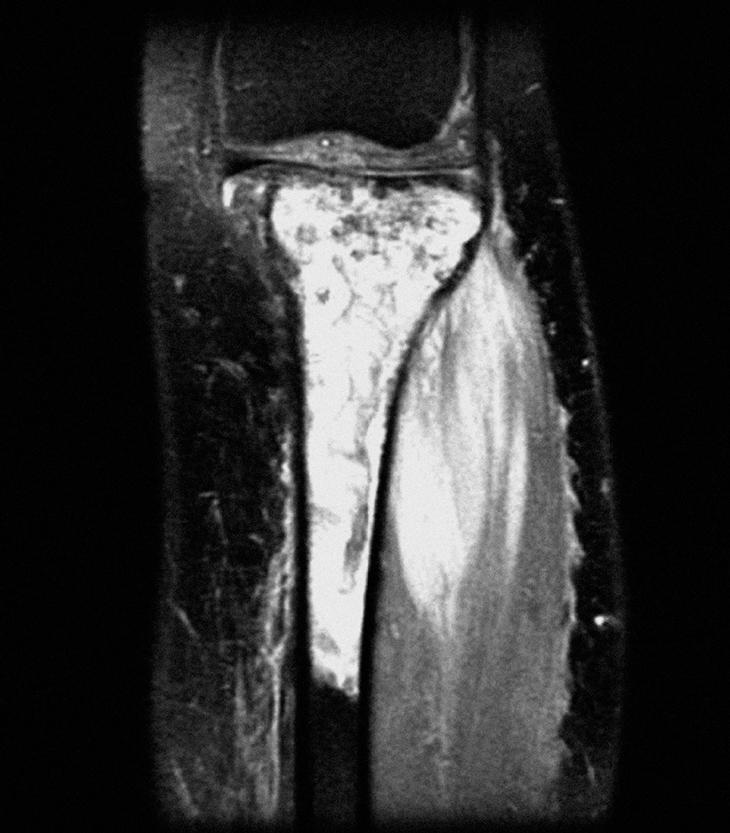
En la radiografía de tibia izquierda, en la metáfisis y diáfisis proximal de tibia (fig. 1) se apreciaron lesiones difusas de márgenes mal definidos con zonas escleróticas y líticas sin reacción perióstica. En el estudio con resonancia magnética (RM) de la tibia se observó una tumoración no expansiva de la mitad superior de la tibia y peroné, con áreas de engrosamiento cortical e infiltración no destructiva, con afección de los músculos tibial anterior y posterior e invasión neurovascular. La lesión no era expansiva pero producía alteración de la cortical con esclerosis reactiva de la zona posterior y masa de partes blandas muy significativa a través de la cortical externa, sin desaparición de ésta, en forma de aspecto infiltrante (fig. 2). Se completó el estudio radiológico con una gammagrafía ósea en la que existía un acúmulo del radiotrazador en la tibia proximal, compatible con reacción osteoblástica, sin captaciones a otros niveles.
Figura 1. Radiografía lateral de la tibia izquierda.
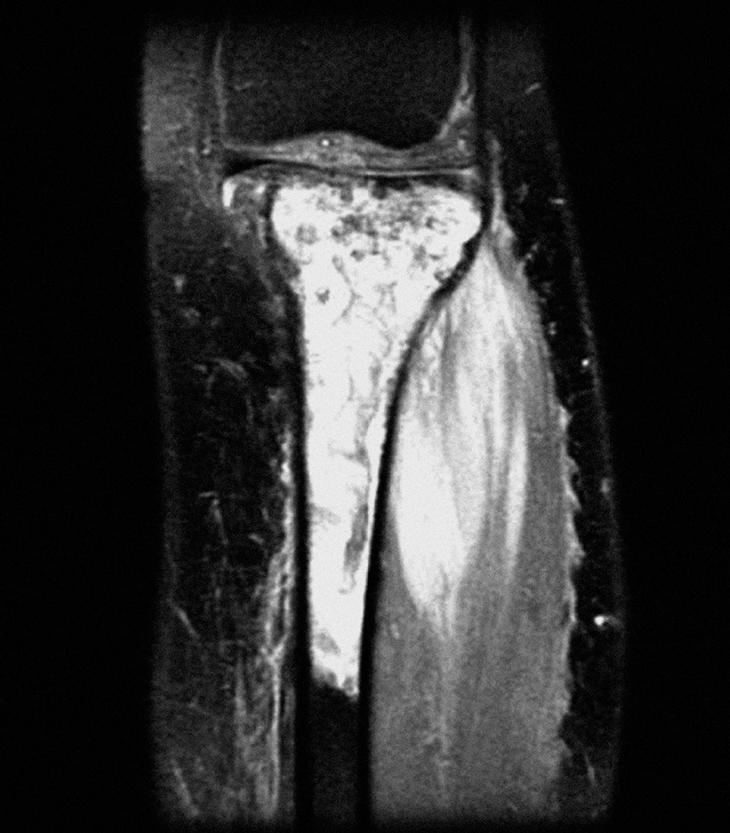
Figura 2. Resonancia magnética coronal STIR de la tibia izquierda, pretratamiento.
Diagnóstico y evolución
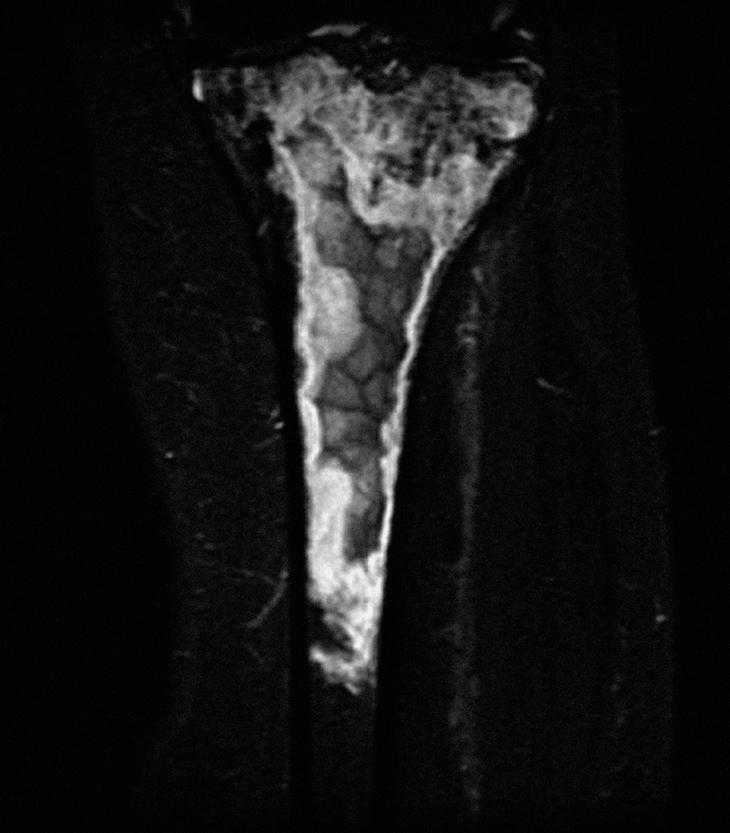
Se realizó una biopsia ósea de la tibia, con el diagnóstico anatomopatológico de linfoma B de células grandes óseo difuso con alto índice proliferativo, según la clasificación de la Organización Mundial de la Salud. En el estudio de extensión con TC toracoabdominopélvico y biopsia de médula ósea, no se observaron alteraciones. Ante el diagnóstico de linfoma óseo primario, la paciente ha recibido tratamiento con quimioterapia con el esquema CHOP + rituximab (6 ciclos) y con radioterapia secuencial posterior. A las 12 semanas, una RM de control mostraba una clara disminución de la afección de partes blandas, pero sin cambios óseos significativos (fig. 3). Desde el punto de vista articular, la paciente se encontraba estable.
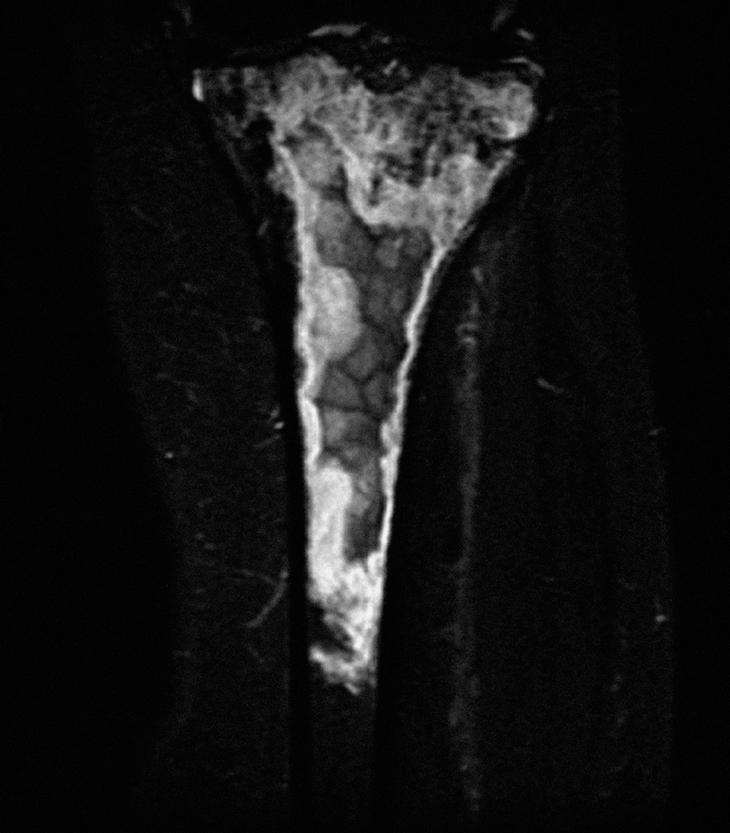
Figura 3. Resonancia magnética coronal STIR de tibia izquierda, postratamiento.
Comentario
El linfoma óseo primario es un linfoma extraganglionar, que comprende un 7% de los tumores malignos óseos y menos del 1% de los linfomas no hodgkinianos1. Puede ser monostótico o poliostótico, con una frecuencia del 66 y 34%, respectivamente. Es más frecuente en varones, con un pico máximo de aparición en la edad adulta. Se localiza principalmente en íleon, columna vertebral, escápula y huesos largos, como el fémur y la tibia. Los síntomas varían según la localización; sin embargo, los más frecuentes son: dolor, tumor palpable o visible e incapacidad funcional del hueso y/o región afectada. El pronóstico depende principalmente del tipo histológico de linfoma, con una supervivencia a los 10 años entre el 60-80%2,3.
Aunque diferentes estudios clínicos no han demostrado un excesivo riesgo de linfoma en pacientes con AR tratados con metotrexato, es cierto que siguen apareciendo casos en los que se describe esta asociación4. En la AR son más frecuentes las formas extraganglionares, sobre todo la localización ósea5. La mayoría de pacientes con enfermedades linfoproliferativas tienen datos de inmunosupresión. De manera que la AR por sí misma y las acciones del metotrexato, pueden concurrir para desarrollar ese estado de inmunosupresión. Los factores de riesgo que desempeñan un papel importante serían: a) la intensa inmunosupresión y la gravedad de la enfermedad en combinación con una susceptibilidad genética, y b) la alta frecuencia de infección latente con prooncogenes del virus de Ebstein-Barr6. En conclusión, el desarrollo de enfermedades linfoproliferativas por tratamiento con dosis bajas de metotrexato, es un tema controvertido. Las características de estas neoplasias y la posibilidad de una remisión después de la suspensión del metotrexato, hacen que no se descarte esta asociación7. Son necesarios estudios epidemiológicos que clarifiquen este tema tan debatido.