


Introducción
La linfadenitis de Kikuchi-Fujimoto (LKF) es una forma rara de linfadenitis necrosante descrita en 1972 por dos anatomopatólogos japoneses. Kikuchi1 describió una linfadenitis caracterizada por una hiperplasia focal de células reticulares con restos nucleares y fagocitosis; Fujimoto describió las características clínicas, y la definió como una linfadenitis cervical necrosante subaguda. Aunque la distribución de la enfermedad en un principio era en países asiáticos, actualmente se conocen casos prácticamente en todo el mundo. Se caracteriza por afectar a adultos jóvenes, generalmente mujeres, en forma de adenopatía cervical dolorosa con fiebre. Esta linfadenitis tiene un curso benigno y autolimitado, y no suele precisar tratamiento. Desde el punto de vista histológico, las alteraciones son muy llamativas, con áreas de necrosis cortical y paracortical, rodeadas de histiocitos, inmunoblastos, linfocitos y monocitos plasmocitoides. Es muy característica la ausencia de polimorfonucleares y la escasez de células plasmáticas (fig. 1)2. La etiopatogenia de esta entidad se desconoce3, aunque se ha descrito en asociación con infecciones y diferentes enfermedades reumáticas autoinmunitarias, entre las que destaca el lupus eritematoso sistémico (LES). El objetivo del presente trabajo es describir las características clínicas de los pacientes con linfadenitis de Kikuchi-Fujimoto e intentar establecer su posible asociación con el lupus eritematoso sistémico.
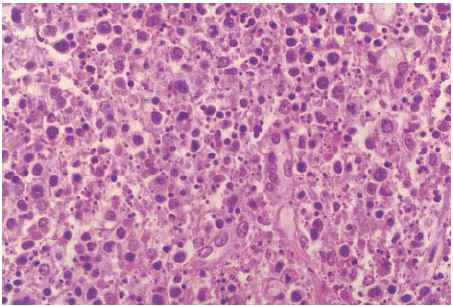
Figura 1. Linfadenitis histiocitaria necrosante. No se evidencian polimorfonucleares pero sí la presencia de una notable cariorrexis (linfadenitis de Kikuchi-Fujimoto) (caso 1) (HE ×400).
Métodos
El diseño fue de tipo retrospectivo. Se utilizó la base de datos de la Sección de Reumatología del Hospital Universitario Germans Trias i Pujol de Badalona. Todos los pacientes estaban codificados según una versión modificada de los códigos diagnósticos del Colegio Americano de Reumatología. Se revisaron las historias clínicas de los pacientes diagnosticados de LES (188) que cumplían los criterios clasificatorios de la American College of Rheumatology (ACR). Asimismo, se revisaron las historias clínicas de aquellos pacientes con el diagnóstico histológico de LKF desde el año 1984 hasta 2001. El Hospital Universitario Germans Trias i Pujol atiende un área de referencia de 800.000 habitantes. En esta área existen 4 servicios de reumatología con un total de 5 reumatólogos.
Resultados
Se encontró 4 mujeres con LKF (media de edad, 31 años; límites, 27-34). En 3 de los 4 casos (75%) la linfadenitis estaba asociada a LES, y en un solo caso la paciente presentó una LKF aislada. A continuación describimos brevemente los cuatro casos de LKF.
Caso 1
Mujer de 34 años de edad, con antecedentes de fotosensibilidad, lesiones cutáneas específicas de lupus eritematoso y artralgias. Presentaba anemia, linfopenia, anticuerpos antinucleares (ANA) positivos a título de 1/320, patrón homogéneo y moteado, y anticuerpos anti-ADN positivos a títulos de 1/1.280. Los anticuerpos anti-Ro, anti-La y anti-Sm también fueron positivos (ELISA). Fue diagnosticada de LES. Un año más tarde consultó por fiebre y adenopatías axilares de 10 días de evolución. Los hemocultivos y las serologías de lúes, toxoplasma, brucela, virus de Epstein-Barr, citomegalovirus y virus de la inmunodeficiencia humana fueron negativos. Se realizó la biopsia de una adenopatía que evidenció una LKF. Con prednisona (1 mg/kg/día por vía oral) la paciente quedó asintomática en pocas semanas.
Caso 2
Mujer de 34 años de edad, con antecedentes de fiebre reumática y valvulopatía mitral, que requirió un recambio protésico a los 16 años de edad. A los 24 años presentó fiebre y adenopatía cervical, que fue biopsiada, llegándose al diagnóstico de LKF. Presentaba unos ANA positivos a título 1/320, patrón homogéneo, con anticuerpos anti-ADN negativos. La paciente no presentaba ninguna otra característica clínica de LES. Se resolvió con prednisona (15 mg/día por vía oral) en pocas semanas. Diez años después presentó una clara fotosensibilidad, aftas orales, anemia, linfopenia y aumento de las cifras de ANA (> 1/2.500). Los anticuerpos anti-ADN eran negativos. Se estableció el diagnóstico de LES.
Caso 3
Mujer de 27 años de edad, que consultó por la presencia de fiebre, sudación nocturna y adenopatías laterocervicales derechas de 2 meses de evolución. En la exploración física presentaba, además, una artritis de rodilla. En el hemograma destacaba una anemia normocítica normocrómica y una linfopenia. El complemento era normal. Se realizó un test de Mantoux que fue negativo. Los hemocultivos y serologías de lúes, brucela, toxoplama, citomegalovirus, virus de Epstein-Barr y virus de la inmunodeficiencia humana resultaron negativos. Los ANA y los anticuerpos anti-ADN también fueron negativos. Se realizó una artrocentesis, obteniéndose un líquido de características inflamatorias con 7.600 leucocitos/μl (40% monocitos), sin cristales. El cultivo en medios ordinarios y la investigación de micobacterias en el líquido articular fueron negativos. Se realizó una biopsia ganglionar que evidenció una linfadenitis necrosante con características de linfadenitis de Kikuchi. Sin embargo, podían observarse detritus de ADN en la pared de los vasos, hallazgo muy sugestivo de linfadenitis lúpica. La paciente quedó asintomática en 3 semanas con tan sólo tratamiento antiinflamatorio para la artritis.
Caso 4
Mujer de 29 años de edad, diagnosticada de LES a raíz de una historia de artritis, leucolinfopenia, ANA positivos 1/2.560 patrón moteado y homogéneo, anti-ADN positivo 1/80 y anti-Ro y anti-Sm positivos. Los anticuerpos anticardiolipina IgG eran positivos. Dos años más tarde, consultó por la presencia de adenopatías laterocervicales dolorosas. La punción-aspiración con aguja fina evidenció una linfadenitis reactiva inespecífica. La tinción de Ziehl-Neelsen fue negativa. La biopsia escisional de una de las adenopatías fue compatible con LKF.
Discusión
La LKF es una forma poco conocida de linfadenitis necrosante. Tiene un curso autolimitado y precisa confirmación histológica para su diagnóstico. Por esta razón, probablemente, está infradiagnosticada. No se dispone de datos de su prevalencia. No obstante, de las principales series publicadas2,4 se deduce una frecuencia que oscila entre el 0,5 y el 5% de todas las biopsias ganglionares realizadas. La serie más amplia corresponde a Dorfman4 et al, con 108 casos diagnosticados en un período de 15 años.
La etiología de la LKF es desconocida y las diferentes teorías son muy controvertidas. Algunos autores proponen una etiología infecciosa, concretamente viral, dado el curso autolimitado y la evidencia simultánea de una infección en algunos casos: parvovirus B195, virus de Epstein-Barr6-9 y herpes virus tipo 610,11. Sin embargo, en los casos en que se han realizado técnicas de PCR e hibridación in situ para intentar demostrar una causalidad de estos virus, los resultados han sido contradictorios12,13. Otros autores, apoyándose en la frecuente asociación con enfermedades autoinmunitarias, especialmente con el LES, postulan un mecanismo inmunitario en la etiopatogenia de este proceso14.
Hasta la fecha se han descrito 28 casos de LKF asociada a LES. Esto representa un porcentaje muy pequeño respecto a la totalidad de casos de LKF. En 13 casos4,15-22 la LKF se presentó antes que el LES; en otros 8 casos5,23-25 el diagnóstico se realizó de forma simultánea, y en 5 casos23,26,27 el diagnóstico de LKF se hizo tiempo después del diagnóstico de LES. En otros 2 casos5 el autor no especifica la cronología de las dos entidades.
Se desconoce si la asociación entre LKF y LES es real o casual. Tampoco se sabe si existe un sustrato fisiopatológico común o simplemente se trata de dos entidades diferentes que se presentan en la misma población. Es fácil aventurar que estamos ante dos entidades diferentes y que simplemente coinciden por mero azar. Sin embargo, es sorprendente que no se haya descrito, por ejemplo, la asociación entre LFK y la artritis reumatoide, por mencionar otra enfermedad reumática prevalente que se presenta en la misma población en cuanto a edad y sexo.
Algunos autores han catalogado a esta linfadenitis como una forma similar al lupus eritematoso sistémico, pero autolimitada5. Y es que la LKF y la linfadenitis lúpica pueden ser histológicamente idénticas. Tan sólo en aquellos pocos casos en que el anatomopatólogo logra observar cuerpos hematoxifílicos (agregados de material basofílico en los senos del ganglio), depósito de ADN en la pared de los vasos o zonas de vasculitis fuera del área de necrosis (fig. 2) sugeriría una linfadenitis lúpica y no una LKF4 (tabla 1).
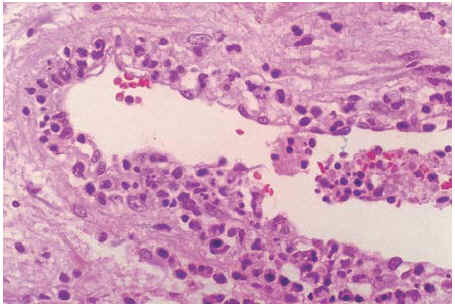
Figura 2. Arteria de mediano calibre del hilio ganglionar, con detritus de ADN en la pared, característica sugestiva de linfadenitis lúpica (caso 3) (HE ×400).

La presentación clínica y la anatomía patológica de los 4 casos aportados apoyan la asociación de la LKF y el LES. Es interesante comprobar cómo en el primer caso una paciente con criterios de LES presenta una linfadenitis histiocitaria necrosante con ausencia de polimorfonucleares típica de LKF. Para algunos autores podría tratarse de una linfadenitis lúpica, puesto que en ocasiones la histología puede ser idéntica y, además, responde al tratamiento con glucocorticoides. En el segundo caso, la paciente presentó una LKF cuando aún no tenía ninguna manifestación de LES, salvo unos ANA positivos a títulos bajos. Unos años más tarde sí cumpliría criterios clasificatorios de LES. En el tercer caso, en cambio, la paciente no presenta ningún signo clínico ni analítico de LES y, sin embargo, la histología ganglionar es compatible con LKF, aunque con algún hallazgo sugestivo de linfadenitis lúpica4. El cuarto caso clínico corresponde a un LES (criterios clasificatorios de la ACR), que 2 años después del inicio de los síntomas presentó una adenopatía cuya biopsia fue compatible con una LKF.
La patogenia o el mecanismo de desarrollo de la LKF se desconoce. Sin embargo, el hecho de que por microscopia electrónica se haya logrado demostrar la presencia de estructuras tubulorreticulares en el citoplasma de linfocitos e histiocitos de ganglios con LFK similares a las observadas en el LES sugiere, como mínimo, una linfadenitis hiperinmunitaria mediada por linfocitos T28.
Tampoco se conoce por qué unos pacientes con LKF evolucionan a LES y otros no lo hacen. No se dispone de ningún dato clínico o analítico que pueda prever la evolución a LES de un paciente diagnosticado de LKF. Recientemente, Arnaudo29 et al comunicaron que en los pacientes con linfadenitis de Kikuchi asociada a lupus los anticuerpos anti-DEK estaban presentes hasta en un 22% de los casos.
Éste podría ser el primer paso para encontrar el nexo común, si existe, entre la LKF y el LES. La descripción de nuevos casos de LKF, asociados o no a LES, y su seguimiento clínico y analítico permitirán establecer mejor la prevalencia de esta asociación, y si se trata o no de diferentes manifestaciones de una misma enfermedad.