



Caso clínico
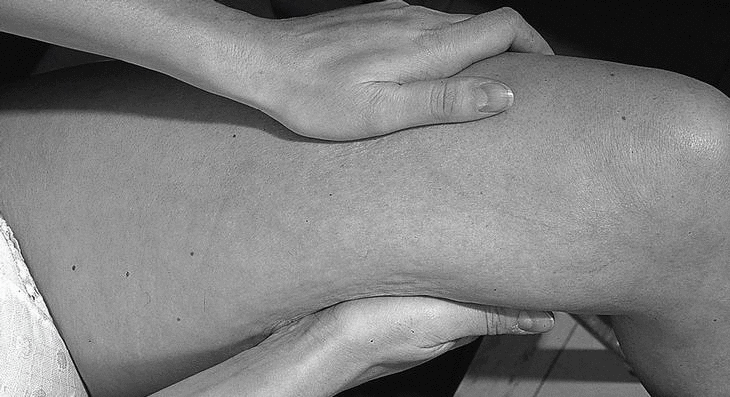
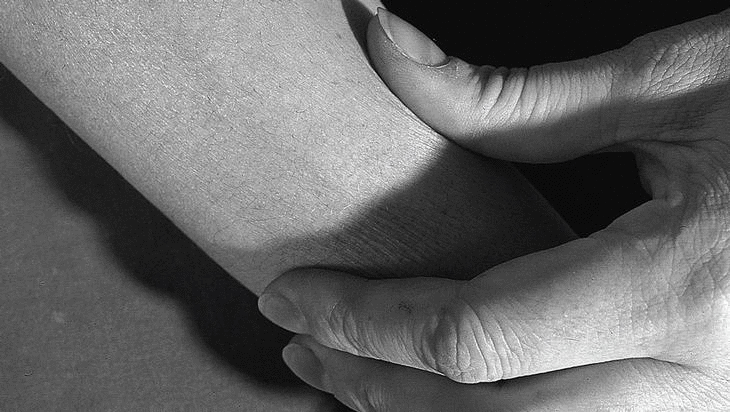
Mujer de 36 años, sin antecedentes de interés, que consulta por presentar durante cuatro meses cansancio y dolores musculares en extremidades, que la paciente describe como «sensación de agujetas» acompañados de edema doloroso, sensación de tirantez en la piel, con aspecto de piel de naranja (fig. 1) y dificultad al pellizcamiento de partes blandas (fig. 2). La exploración física mostraba una discreta limitación dolorosa de la movilidad de las articulaciones (muñecas, codos y rodillas) así como edema que dejaba fóvea evidente con pérdida de los pliegues cutáneos en antebrazos, manos y piernas. Refería disminución de fuerza en todos los grupos musculares de las extremidades, sin otros hallazgos valorables. En los análisis de sangre destacaba una elevación de los reactantes de fase aguda (VSG 60 mm/primera hora, proteína C reactiva 83,2 μg/dl), una eosinofilia del 23%, con anemia ferropénica e hipercomplementemia C4 (56 mg/dl), elevación de las α y β-globulinas en el proteinograma y valores normales de las enzimas musculares. El EMG mostraba una afección miógena grave y generalizada en miembros, preferentemente distal. El rastreo óseo, las radiografías de manos, pelvis, rodillas y pies estaban dentro de la normalidad. Se practicó una biopsia de piel y músculo en el antebrazo cerca del codo.
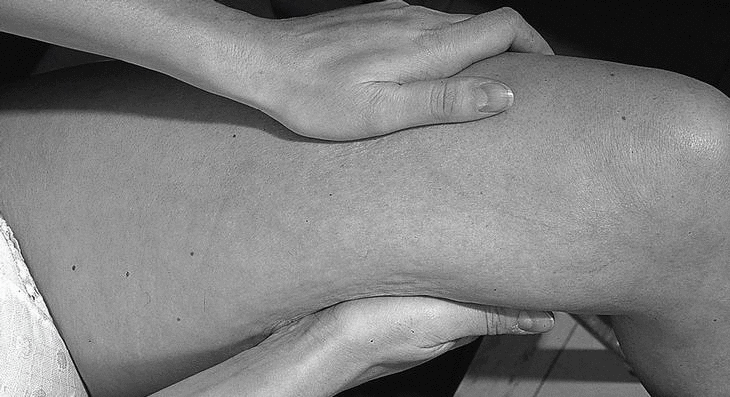
Figura 1. Cara interna de muslo con aspecto de piel de naranja.
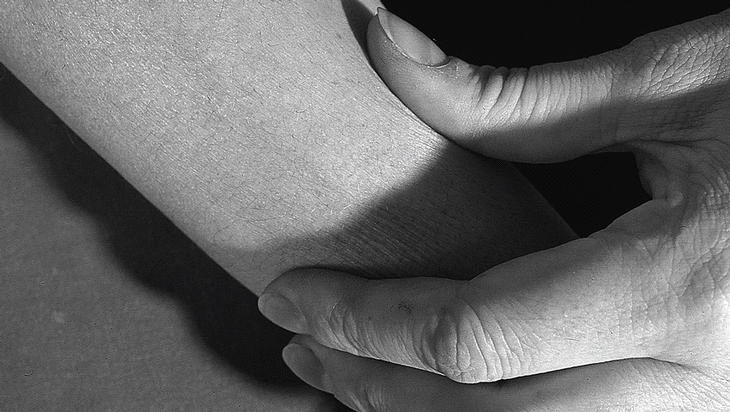
Figura 2. Dificultad al pellizcamiento de partes blandas en la cara anterior del antebrazo.
Diagnóstico y evolución
La biopsia mostró una afección inflamatoria importante de la fascia, con predominio eosinófilo, y discreta alteración muscular, confirmándose el diagnóstico de fascitis eosinófila (fig. 3). Se inició tratamiento con corticoides a dosis de 1 mg/kg/día, obteniendo una mejoría ostensible.

Figura 3. Biopsia: músculo rojizo y fascia engrosada teñida de azul, con escasa celularidad y surcada por infiltrado inflamatorio. (Tricrómico de Masson, 40 x 20.)
Comentario
La fascitis eosinófila (FE), descrita por Shulman en 19741, es una enfermedad caracterizada por eosinofilia periférica, hipergammaglobulinemia y fascitis difusa con ausencia de manifestaciones sistémicas. Es más frecuente en varones, entre la tercera y sexta décadas de la vida; en algunas series la incidencia es la misma para ambos sexos2.
Suele presentarse de forma aguda o subaguda con dolor, edema e hipersensibilidad que posteriormente da lugar a placas duras y dolorosas, localizadas con mayor frecuencia en antebrazos y pantorrillas de las extremidades, como ocurrió en esta paciente. Las zonas afectadas pueden adquirir aspecto de piel de naranja o de empedrado con preservación de la epidermis. Puede acompañarse de deformidades articulares (55-75%), artritis no erosiva (40%), placas de morfea (25-30%)3 y síndrome del tunel carpiano (20%). La afección visceral en la FE es muy poco frecuente2.
En el hemograma destaca la eosinofilia, presente ya en las fases iniciales, llegando a cifras superiores a 1.000 por µl que tienden a normalizarse con el tratamiento con corticoides2, aumento de los reactantes de fase aguda e hipergammaglobulinemia policlonal, que puede aparecer hasta en el 75% de los casos. El diagnóstico certero de la FE requiere la biopsia de la zona afectada que incluya la piel y el músculo superficial. El examen histológico muestra un engrosamiento de la fascia, adherida al músculo adyacente, con gran infiltrado inflamatorio. No se considera necesaria la presencia de eosinófilos para realizar el diagnóstico3, pero parece haber una estrecha correlación entre la eosinofilia sanguínea y la tisular2. La inflamación también se extiende a los septos fibrosos de los lóbulos grasos de la hipodermis. En fases avanzadas, se produce una regresión espontánea del infiltrado celular, que es sustituido por fibrosis4.
Clínicamente, el diagnóstico diferencial se plantea con colagenopatías como la esclerodermia sistémica, la dermatomiositis, la artritis reumatoide o ciertas paniculitis. Inicialmente, en nuestra paciente sospechamos una dermatomiositis, aunque llamaba la atención una afección más distal de las extremidades, menos frecuente en esta enfermedad. El número tan elevado de eosinófilos nos obligó a pensar en otros procesos que cursan con hipereosinofilia, como la vasculitis de Churg-Strauss y el síndrome de hipereosinofilia idiopática; así como con el síndrome de eosinofilia miálgica (SEM) y el síndrome producido por el aceite tóxico (SAT)2 que en algún momento de su evolución presentan cambios esclerodermiformes. En los dos primeros no hay fascitis ni afección muscular, mientras que en los dos últimos (SEM y SAT) predominan las mialgias y las alteraciones se extienden a otros órganos y tejidos6-8. La bioquímica descartó la dermatomiositis; no fue la biopsia la que confirmó el diagnóstico.
El pronóstico de la FE es bueno, generalmente autolimitado con una duración de 1 a 3 años, aunque a veces recidiva o se cronifica. El tratamiento recomendado es la corticoterapia a dosis de 40-60 mg/día de prednisona repartidos en 2-3 tomas, reduciéndola progresivamente según se produzca la mejoría y manteniéndola de 6 a 12 meses tras la remisión5, junto con fisioterapia. Puede asociarse hidroxiclorocina (200-400 mg/día), y la respuesta a este tratamiento es muy variable9. En los casos rebeldes y para reducir las dosis de corticoides, se propone el uso de metotrexato semanal.