


Introducción
Desde que Bowie et al describieron un grupo de pacientes con lupus eritematoso generalizado (LEG) y fenómenos trombóticos a pesar de tener anticoagulante lúpico circulante1, nació el interés sobre esta paradoja. Veinte años después, Harris et al informaron de que los fenómenos trombóticos en pacientes con LEG estaban asociados con anticuerpos anticardiolipina (ACL)2. A esta asociación se le denominó inicialmente «síndrome de anticardiolipina»3, pero cuando se supo que esos anticuerpos reconocían también a otros fosfolípidos (AAF), el nombre se amplió a «síndrome de antifosfolípidos» (SAF). A la trombosis se le agregó pérdidas fetales recurrentes, trastornos neurológicos, trombocitopenia, anemia hemolítica y livedo reticularis como componentes del SAF. Si éste se asocia a una enfermedad autoinmunitaria, normalmente LEG, se le conoce como SAF secundario4 y si ocurre en pacientes sin ninguna enfermedad autoinmunitaria asociada se le llama SAF primario5-7.
En 1990, tres grupos informaron que los AAF de pacientes con SAF requerían de una proteína, identificada como β2-glucoproteína-I (β2GP-I), para su detección in vitro8-10. Ésta es una proteína sérica presente en todos los individuos sanos que interviene en algunos procesos de coagulación, aterogénesis y apoptosis. Por ello, el estudio de la β2GP-I en la patogenia del SAF ha despertado gran interés.
Aquí revisamos algunos aspectos relacionados con la participación del sistema β2GP-I/anti-β2GP-I en la patogenia del SAF y como exordio para el análisis del potencial que ofrece el estudio del polimorfismo de la β2GP-I, para entender mejor su participación en el desarrollo del síndrome de antifosfolípidos tocamos algunos puntos relevantes de la estructura de la también llamada apolipoproteína H.
Síndrome de antifosfolípidos
El SAF es una enfermedad autoinmunitaria considerada como el resultado de la interacción de factores ambientales, hormonales, inmunorreguladores y genéticos. Actualmente, se utiliza el término «síndrome de antifosfolípidos» para definir la entidad clínica asociada con la presencia de anticuerpos antifosfolípidos (AAF) con trombosis arteriales y/o venosas, abortos de repetición, livedo reticularis, trombocitopenia, anemia hemolítica y alteraciones neurológicas11. La asociación de estas manifestaciones clínicas con los AAF fue reportada originalmente en pacientes con LEG; sin embargo, hay pacientes con títulos elevados de AAF y las manifestaciones clínicas anotadas, pero sin datos clínicos ni serológicos de otra enfermedad autoinmunitaria. A esta entidad clínica se la denomina «síndrome de antifosfolípidos primario» (SAFP), para diferenciarla de la que se asocia con otras enfermedades del tejido conjuntivo, principalmente LEG4. Existen diferencias entre ambas entidades, debidas principalmente a la presencia de valores más elevados de AAF en el SAFP y a la influencia de LEG en el síndrome de antifosfolípidos secundario (SAFS)5.
Anticuerpos antifosfolípidos
Los AAF son una familia de inmunoglobulinas cuyo origen puede estar determinado por los siguientes factores: a) autoanticuerpos naturales12,13 presentes en todos los mamíferos a títulos relativamente bajos, codificados por genes de línea germinal; b) resultado de la influencia de factores inmunogenéticos14; c) desregulación inmunitaria con excesiva producción de autoanticuerpos15; d) parte de la red de anticuerpos antiidiotipo16-18; e) inducidos por fármacos (p. ej., clorpromazina)19, y f) resultado de procesos infecciosos20.
Factores proteicos asociados al SAF
El año 1990 marca un hito en el estudio de los AAF. En ese año, la investigación en el campo de los AAF cambió de rumbo debido a que tres grupos reportaron simultáneamente que los AAF requerían una proteína con afinidad por estructuras aniónicas para su detección in vitro, identificada como β2GP-I8-10. Estudios posteriores señalaron que algunos autoanticuerpos contra fosfatidiletanolamina también requieren cofactores proteicos21. Recientemente, se ha reportado que los «anticuerpos antifosfolípidos» pueden reaccionar contra una gran variedad de proteínas plasmáticas, como protrombina, proteína C, proteína S, quininógeno y antitrombina III, y otras proteínas unidoras de fosfolípidos, como la anexina V 22-25.
A partir de la publicación de estos hallazgos, los inexactamente llamados cofactores han tenido un papel protagonista dentro del SAF. La denominación cofactor se acuñó desde los primeros estudios que demostraron la participación de proteínas plasmáticas en el reconocimiento de los AAF; sin embargo, actualmente se acepta que los autoanticuerpos asociados al SAF reconocen epítopos localizados propiamente en las estructuras proteicas nativas o bien asociadas a fosfolípidos; por ello, consideramos que la denominación propuesta en principio es inexacta.
De los factores proteicos asociados al SAF, la β2GP-I es el autoantígeno más estudiado y los anticuerpos anti-β2GP-I son los que con mayor frecuencia están asociados a este proceso. Uno de los fosfolípidos con el que la β2GP-I forma un complejo antigénico es el difosfatidilglicerol, fosfolípido aniónico comúnmente conocido como cardiolipina (CL). De acuerdo con lo expuesto anteriormente, en un ensayo convencional para detectar anticuerpos anticardiolipina (ACL), en el que se usa suero bovino fetal (contiene β2GP-I) como bloqueador, la unión de los ACL puede atribuirse a epítopos nativos de la β2GP-I9,11, al complejo β2GP-I-CL8, o al reconocimiento de epítopos crípticos en la CL26 y/o en la β2GP-I27. Por ejemplo, con espectroscopia infrarroja27 y dicroísmo circular28 se demostró que la interacción de la β2GP-I con la CL produce cambios conformacionales importantes en ambas moléculas, generando probablemente una exposición de regiones crípticas. Los resultados de los estudios revelaron que las alteraciones en la molécula fosfolipídica restringen la movilidad de las cadenas hidrocarbonadas, mientras que la β2GP-I sufre modificaciones en su estructura que disminuyen el porcentaje de arreglos α-hélice y β-plegada, e incrementan la conformación azarosa, aumentando así su potencial antigénico. Estos datos sugieren que los epítopos crípticos detectados por los ACL se encuentran probablemente en la β2GP-I modificada por su unión a la cardiolipina. Por otro lado, este neoepítopo no sólo puede ser el resultado de la unión con la cardiolipina ya que Matsuura et al demostraron que ese mismo cambio conformacional puede ser inducido cuando la β2GP-I entra en contacto con superficies oxigenadas29, es decir, con una densidad electrónica intrínsecamente elevada. Ésta es quizá la razón por la que los ACL y los anti-β2GP-I detectados en placas irradiadas tienen un alto coeficiente de correlación; por ello, actualmente se considera que el ELISA convencional para ACL detecta, además de los AAF verdaderos, los anticuerpos anti-β2GP-I. Alternativamente, nuestro grupo ha propuesto que los anti-β2GP-I en placas no irradiadas detectan un epítopo diferente presente en la proteína nativa y que estos anticuerpos se asocian con más fuerza al SAF que los mismos ACL11.
Participación de la β2GP-I en el SAF
Aunque Galli et al9 y Cabral et al30 ya habían informado sobre la existencia de anticuerpos anti-β2GP-I libre de fosfolípidos en pacientes con SAF, Viard et al informaron por primera vez de la asociación de trombosis con anti-β2GP-I en pacientes con LEG31. Posteriormente, Gharavi, en 199232, indujo SAF experimental mediante la inmunización de ratones con β2GP-I. En 1995, Cabiedes et al reportaron que los anti-β2GP-I se asocian con mayor fuerza a las manifestaciones trombóticas del SAF y al SAF mismo que los ACL propiamente dichos33. Un metaanálisis posterior confirmó estos resultados34. Estos y otros reportes confirmatorios han señalado la gran relevancia que tiene el sistema β2GP-I/anti-β2GP-I en la patogenia del SAF35-43.
Bioquímica de la β2GP-I
Descrita en 196144, la β2GP-I humana, también llamada apolipoproteína H, pues activa la lipasa de lipoproteína45,46, es una proteína presente en el plasma de todas las personas sanas a una concentración aproximada de 200 μg/ml. Está constituida por una sola cadena polipeptídica de 326 aminoácidos47-49, pesa 50 kD y aproximadamente el 18% de su peso está constituido por carbohidratos. Su gen reside en el cromosoma 17q23-qter50. La β2GP-I está organizada en cinco dominios homólogos conocidos como segmentos cortos repetidos (short consensus repeats, SCR)51, cada uno tiene aproximadamente 60 aminoácidos con puentes disulfuro inter e intrarregiones52. El sitio de unión a fosfolípidos de carga negativa reside en el quinto SCR en la zona que involucra a los aminoácidos 281-288 (CKNKEKKC)53, 54; dado su alto contenido de aminoácido básico lisina (K), a pH fisiológico dicha secuencia es catiónica.
Papel fisiológico y patogénico de la β2GP-I
La β2GP-I inhibe la activación por contacto de la vía intrínseca de la coagulación55, la agregación plaquetaria mediada por ADP56, la generación de trombina55, la actividad del complejo de la protrombinasa56 y la activación del sistema de la proteína C57,58. Por estas propiedades in vitro se ha propuesto a la β2GP-I como anticoagulante natural. Por la inducción de aterosclerosis experimental y otros hallazgos similares, se ha propuesto que la β2GP-I tiene un papel preponderante en la regulación del metabolismo de los lípidos59,60. De acuerdo con esto, las trombosis de repetición características del SAF inducidas por los anti-β2GP-I pueden ser el resultado de una inhibición de la función de su antígeno o, menos probablemente, una disminución de su concentración plasmática61,62. Otras propuestas son el depósito de complejos inmunitarios en la pared de los vasos, y la activación mediada por células (p. ej., el incremento de la adhesión de monocitos al endotelio)63.
β2GP-I: aspectos genéticos
La estructura molecular de la β2GP-I, como la de todas las proteínas, está determinada genéticamente y hay variantes cuantitativas y cualitativas entre los individuos de los diversos grupos étnicos. La variación cuantitativa es el resultado de la presencia de un alelo nulo (Bg*D) que es codominante con respecto al alelo normal (Bg*N)64. La frecuencia del alelo nulo es alta (0,26) en la población negra de Mocambique (tabla 1) y en las poblaciones asiáticas de Irán, Afganistán y Corea (0,11, 0,12 y 0,22, respectivamente) y baja en la población caucásica (aproximadamente de 0,06)65. Los individuos homocigotos para el alelo Bg*D presentan valores séricos normales de β2GP-I (160-300 μg/ml), en tanto que en los heterocigotos sus valores son bajos (60-140 μg/ml). Es interesante que, los valores plasmáticos de la β2GP-I en los individuos homocigotos para el alelo deficiente están por debajo de los límites de detección66. A pesar de lo anterior, es importante señalar que, además del factor genético, el sexo en los individuos caucásicos también influye en la variación de la concentración de β2GP-I. En este grupo étnico los valores plasmáticos de β2GP-I son menores en mujeres67.

Además de la variación cuantitativa, existen diferencias estructurales de la β2GP-I. Kamboh et al estudiaron estas diferencias en la población estadounidense de razas negra y blanca mediante isoelectroenfoque (IEE), seguido de inmunoprecipitación revelada con un antisuero policlonal para identificar la β2GP-I66. La investigación demostró que la variación estructural era de dos tipos: intraindividual, también conocida como microheterogeneidad, la cual es el resultado del grado de glucosilación de la proteína, e interindividual, ocasionada por sustituciones de aminoácidos, es decir, determinada genéticamente. De esta última variación ha sido posible detectar cuatro variantes alélicas, designadas como APOH*1, APOH*2, APOH*3 y APOH*4. El alelo APOH*4 se encuentra únicamente en personas de raza negra de los EE.UU., Nigeria y Australia68-72. Los datos obtenidos en el estudio de familias condujeron a establecer que el patrón de herencia seguido por estos alelos es autosómico codominante.
El polimorfismo estructural ha sido estudiado en otras poblaciones de los cinco continentes, en donde se ha encontrado una frecuencia predominante del alelo APOH*2, por lo que es considerado el alelo silvestre. Cabe resaltar que existe una cierta similitud en la distribución de frecuencias entre las diversas poblaciones que pertenecen a la misma raza. En el caso de la raza negra la homogeneidad entre poblaciones evidenció una disminución en la frecuencia del alelo APOH*1. En contraste con esto, en los aborígenes australianos se presentó un incremento importante en la frecuencia del alelo APOH*1 (0,13, tabla 2) y una ausencia del alelo APOH*363. Por último, en la población de Siberia se encontraron las frecuencias mas bajas de APOH*2 y las más altas de APOH*373. En la tabla 2 se exponen las frecuencias encontradas hasta 199974,75.

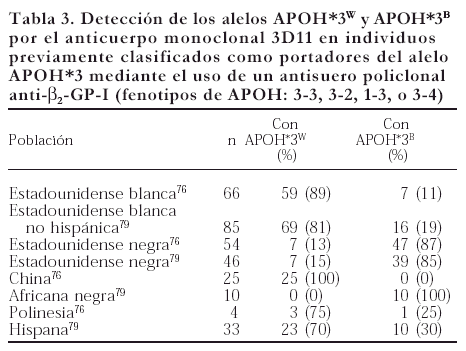
En 1995 se reportó que el alelo APOH*3 tiene dos variantes: APOH*3w y APOH*3B. Lo anterior tiene como sustento el hecho de que la proteína producto de este alelo puede o no ser reconocida por el anticuerpo monoclonal 3D11 anti-β2GP-I76. El 3D11 sólo reconoce el producto del alelo APOH*3 y presenta reactividad preferentemente por la proteína de la raza blanca, por lo que al alelo que codifica para la β2GP-I reconocida por este anticuerpo se le denominó APOH*3w (W, del inglés white), mientras que al alelo del producto no reconocido se le llamó APOH*3B (B, del inglés black) por presentarse predominantemente en individuos de raza negra76. En la tabla 3 se incluye la frecuencia de las variantes del alelo APOH*3 en diferentes poblaciones. Un hallazgo interesante de este estudio es que la β2GP-I de algunos individuos homocigotos para el alelo APOH*3 no une fosfolípidos aniónicos (CL o fosfatidilserina), es decir, no presenta modificaciones estructurales al unirse a superficies aniónicas, lo cual sugirió que este fenómeno es consecuencia de la presencia homozigota del alelo APOH*3w.
Bases moleculares de los polimorfismos estructurales
Las variaciones cuantitativas y cualitativas de la β2GP-I debidas a la existencia de diferentes alelos presentes en un individuo son el resultado de variaciones en el ADN. En 1998 Yasuda et al reportaron el caso de una paciente de 36 años de edad con deficiencia de β2GP-I77. El análisis del gen de la β2GP-I demostró que el exón 4 carecía de la timina correspondiente a la posición 379 del ADN complementario (ADNc). Consecuentemente, se presentaba un corrimiento en el marco de lectura que ocasionaba la aparición de un codón de terminación en el exón 6. Esta mutación en el exón 4 podría ser la base molecular de la presencia del alelo nulo; es decir, la justificación del polimorfismo cuantitativo. De lo anterior puede colegirse que algunas mutaciones puntuales pueden ocasionar variaciones cuantitativas de la β2GP-I. Otras, en cambio, alteran la información del codón correspondiente, resultando en el reemplazo de un aminoácido por otro71,78,79. Los polimorfismos reportados por esta razón se encuentran en las posiciones 88, 247, 306 y 316 de la β2GP-I. En la tabla 4 se indican las posiciones de estas mutaciones y las enzimas de restricción empleadas para detectarlas.

Existen datos experimentales que han demostrado que la mutación en la posición 88 es específica para el alelo APOH*173. Esta mutación, que sustituye serina por asparagina, estuvo presente en todos los portadores del alelo APOH*1 del estudio realizado por Sanghera et al; es decir, parece distinguir el alelo APOH*1 del silvestre APOH*271. En este mismo estudio se encontró que la mutación en la posición 316 (triptófano/serina) establece la diferencia entre los alelos APOH*3W y APOH*2. Es oportuno señalar que, a pesar de los resultados de esta investigación, no se descarta que los alelos APOH*1 y APOH*3W puedan tener otras mutaciones73.
Aún no se ha encontrado relación alguna entre el polimorfismo en la posición 247 y los alelos reportados para las variaciones cualitativas, lo que probablemente indica que este polimorfismo es responsable de la presencia de otras variantes78,79. Esta conjetura la basamos en el siguiente razonamiento: el punto isoeléctrico depende de los pka's de los aminoácidos que constituyen cada proteína; sin embargo, si se considera que los dos aminoácidos involucrados en el polimorfismo 247 (valina y leucina) sólo difieren estructuralmente en un metileno (-CH2-), esto nos sugiere que los puntos isoeléctricos de las dos posibles variantes fenotípicas son tan cercanos que el IEE es incapaz de distinguirlos.
De acuerdo con las mutaciones puntuales reportadas para el gen de la β2GP-I, teóricamente existen 16 polimorfismos proteicos potenciales si consideramos las posibles combinaciones de las cuatro mutaciones. La reclasificación del alelo APOH*3 en dos diferentes alelos y la ausencia de correlación entre las variantes estructurales y el polimorfismo en la posición 247 ponen de manifiesto que la técnica empleada (IEE) en el estudio de las variantes alélicas de la β2GP-I, por su baja resolución, no garantiza la detección de todas las posibilidades reales. Por esta razón, el análisis del polimorfismo de la β2GP-I y sus posibles asociaciones con ciertos procesos patológicos o manifestaciones clínicas es mejor cuando se estudia el ADN68-72.
Relevancia del polimorfismo de la β2GP-I en la patogenia del SAF
El análisis de las familias de pacientes con SAF ha demostrado que el factor genético contribuye a expresar la enfermedad. Por ejemplo, un estudio retrospectivo demostró que el 41% de los pacientes con SAFP y el 35% de los pacientes con SAFS tenían uno o más familiares con evidencias de al menos una manifestación clínica del SAF (p. ej., trombosis o pérdida fetal recurrente)80. Los familiares afectados fueron principalmente mujeres (madre, abuela, hermana), lo que concuerda con la tendencia de género presente en el SAF. Varios grupos han demostrado que los pacientes con SAF tienen mayor frecuencia de DR7 o DR8, según sea el grupo étnico estudiado81,82.
El quinto dominio de la β2GP-I tiene importancia patogénica pues ahí está el sitio que une fosfolípidos53,54,83. En 1998, se observó que algunas variaciones en el gen de la β2GP-I podían afectar su unión a FL aniónicos (p. ej., CL o fosfatidilserina) y consecuentemente su reactividad con los ACL. Las mutaciones responsables del fenómeno se localizaron en las posiciones 306 (cys-gly) y 316 (trp-ser). La β2GP-I de los individuos homocigotos para cada mutación o los heterocigotos para ambas no unió FL aniónicos84, fenómeno que probablemente se debe a que el tipo silvestre codifica para triptófano en la posición 316 y, al parecer, la mutación Ser316 afecta la unión del FL, ya que involucra la inserción de un aminoácido polar (serina) en la secuencia 313-316 (leu-ala-phe-trp) que, posiblemente, interviene en las interacciones no polares entre las dos moléculas. En el caso del polimorfismo 306, el fenómeno puede deberse a una alteración de la configuración normal del quinto dominio de la β2GP-I, debido a que la mutación ocasiona la sustitución de cisteína, el cual forma un puente disulfuro con su homólogo de la posición 281, posición vecina al sitio de unión a FL (281-288).
Según se ha mencionado, la unión de la β2GP-I a los FL aniónicos es esencial para su reactividad con algunos de los ACL presentes en sujetos con SAF; así, la variación genética de la β2GP-I que impide la unión de los FL con la proteína consecuentemente afectará la producción de ACL. Este fenómeno fue examinado en 222 pacientes con LES, comprobándose que la distribución del polimorfismo 316 trp/ser fue significativamente diferente entre los pacientes positivos para ACL de los pacientes sin ACL, lo cual sugiere una probable protección contra la producción de ACL por parte del alelo mutante en la posición 316. A diferencia de esto, la presencia de anti-β2GP-I no se asoció con ningún polimorfismo en pacientes con SAF85. Otros estudios86-88 han sugerido que los autoanticuerpos presentes en pacientes con SAF también reaccionan con el dominio I de la proteína. Es decir, hay anti-β2GP-I que reconocen la proteína nativa puesto que la presencia del fosfolípido no modifica su reconocimiento.
Por otro lado, el cuarto dominio también puede contener un epítopo críptico para los ACL88-91. Mediante el uso de mutantes de β2GP-I y con el anticuerpo monoclonal ACL EY1C889, dicha región fue localizada en la cara interna del dominio IV, normalmente escondida por el dominio III90, la cual puede exponerse al modificar tres interacciones electrostáticas entre el dominio IV y el V91. Estas interacciones electrostáticas que mantienen unidos a los dos dominios son puentes de hidrógeno entre un aminoácido del dominio IV y un aminoácido localizado en el dominio V92. Los puentes de hidrógeno se localizan entre los aminoácidos: D193-K246, D222-K317 y E228-K308.
Hasta la fecha, no se ha identificado ninguna mutación puntual en el cuarto dominio; sin embargo, el polimorfismo de la posición 247 se encuentra entre el sitio unidor de fosfolípidos y la región que expone el epítopo críptico en el dominio IV. Esta condición, sumada a la observación de que la posición 246 establece un puente de hidrógeno con el dominio IV en la estructura terciaria de la β2GP-I, hace suponer que la alteración ocasionada con la sustitución de un aminoácido en la posición vecina provocaría un cambio conformacional posiblemente relevante en el reconocimiento tanto de los ACL como de los anti-β2GP-I, cuyos epítopos se localizarían en los dominios V, III y IV91.
El polimorfismo en la posición 247 ha sido estudiado en pacientes con SAF de las poblaciones asiáticas93, caucásicas93,94, mestiza mexicana95 y negra americana93. Destaca que, a diferencia del polimorfismo estructural, el polimorfismo en la posición 247 tiene una distribución heterogénea entre las distintas poblaciones (tabla 5).

En 1999, un estudio de pacientes asiáticos con SAFP y SAFS considerados como una población homogénea determinó que la presencia del alelo valina y el genotipo val/val están asociados significativamente a la presencia de anti-β2GP-I93 (p = 0,0018 y p = 0,0003, respectivamente). En este mismo estudio, Hirose et al no encontraron asociación alguna con las poblaciones negra y caucásica. Sin embargo, Atsumi et al informaron sobre la existencia de una correlación entre el alelo valina y los pacientes caucásicos con SAFP y ACL positivos94. En este último estudio también se analizaron los polimorfismos en las posiciones 88 y 316: ambos carecieron de correlación con la enfermedad o la presencia de ACL. Otro dato importante de esta investigación fue el incremento del alelo leucina en el grupo de pacientes con SAFS y ausencia de ACL. El ensayo de ELISA para detectar los ACL en este estudio utilizó placas irradiadas, por lo que es posible que en estos pacientes no fuera posible la detección de otros AAF. Es importante destacar que Hirose et al no analizaron a los pacientes con SAF por separado (SAFP y SAFS), además de que la población caucásica estudiada correspondía a individuos de Norteamérica, mientras que Atsumi et al realizaron su estudio con una cohorte de población inglesa.
En la población mestiza mexicana no hubo correlación entre el polimorfismo en la posición 247 y el SAFP; sin embargo, el genotipo leu/leu estuvo significativamente aumentado en pacientes con SAFS95. En resumen, los datos demuestran que en pacientes asiáticos con SAF y caucásicos con SAFP el alelo valina se relaciona con la generación de β2GP-I antigénica, por lo que se podría considerar un factor de riesgo en estas poblaciones; mientras que en pacientes mestizos mexicanos con SAFS, la presencia de anti-β2GP-I se asoció con el genotipo leu/leu. Independientemente del polimorfismo de la β2GP-I, su estructura está determinada genéticamente, por ello debería existir una tolerancia inmunológica para cualquier variante individual. Queda entonces por dilucidar por qué la presencia de cualquiera de los dos aminoácidos en la posición 247, así como el posible cambio conformacional que ocasionan, se asocia con una respuesta autoinmunitaria anormal. Por tanto, parece necesaria la participación de otros factores, probablemente relacionados con la reactividad de las células T, que podrían ser críticos en la pérdida de la tolerancia. Posiblemente, en algunos individuos en los que la susceptibilidad genética depende de algún genotipo en particular, la presencia de infecciones permitiría el procesamiento de proteínas exógenas con epítopos similares a los de la β2GP-I, estimulando así a las células T autorreactivas. Este mecanismo ya ha sido documentado en un modelo murino96. Además, algunos procesos infecciosos se han asociado con la etiología del SAF97-100. Shoenfeld et al, en 1998, informaron de la presencia de secuencias peptídicas de los dominios I, III y IV en virus, bacterias y parásitos97. Más aún, algunos péptidos sintéticos análogos al sitio de unión de los fosfolípidos homólogos en algunos virus, al ser inoculados en ratones, han inducido la producción de ACL y anti-β2GP-I98.
Conclusiones
El SAF es el resultado de la interacción de factores genéticos, ambientales e inmunológicos. El grado de contribución de cada elemento es aún incierto. Independientemente de esto, el análisis del polimorfismo de la β2GP-I ha generado interés porque parece tener cierta relevancia en la generación de los AAF asociados al SAF. Además, su estudio ha permitido una mejor comprensión de la participación de la β2GP-I en este proceso patológico. Por ejemplo, hay datos experimentales que sugieren un posible papel protector de los polimorfismos en las posiciones 306 y 316 contra la producción de AAF. Por otro lado, el polimorfismo en la posición 247 de la β2GP-I ha sido involucrado en la producción de autoanticuerpos propios del SAF; no obstante, la asociación con un determinado alelo ha dependido de la población estudiada y de la variante de SAF analizada. Es importante considerar que, debido a la complejidad de la enfermedad, la cantidad de pacientes diagnosticados con SAF, el reducido número de estudios en este aspecto y el valor inferencial de éstos, cuyo significado estadístico sólo es válido para cada muestra estudiada, es difícil ponderar con precisión el papel del polimorfismo en la posición 247 de la β2GP-I.
Si consideramos la teoría del mimetismo molecular y la posible participación de las infecciones en la etiología del SAF, las diferencias estructurales de la β2GP-I, especialmente las referentes al polimorfismo en la posición 247 y sus consecuencias en la producción de los autoanticuerpos presentes en el SAF, adquieren gran relevancia. Finalmente, consideramos que con el estudio del polimorfismo de la β2GP-I se obtendrán datos que permitirán aclarar la aún incierta participación de los factores genéticos en la etiología y la patogenia del síndrome de antifosfolípidos.
Agradecimientos
El presente trabajo fue apoyado por el Consejo Nacional de Ciencia y Tecnología, México (Nº 34750-N). El Dr. Cabiedes recibe apoyo económico de la Fundación Teletón México, para el estudio de los anticuerpos antifosfolípidos/cofactores y temas afines. El Dr. Cabral recibe apoyo económico del fondo Beatriz Vázquez Samano para la investigación del síndrome de antifosfolípidos/cofactores.