


Una enfermedad recién descrita
El reconocimiento de la heteroplasia ósea progresiva (HOP) como una entidad independiente se produjo en 19941, gracias a la sagacidad del Dr. Fred Kaplan. La oportunidad de revisar más de 125 casos de miositis osificante progresiva, más recientemente denominada fibrodisplasia osificante progresiva (FOP), una enfermedad poco frecuente ocasionada por un aumento en la expresión de la proteína ósea morfogenética 42,3, le permitió observar que algunos pacientes presentaban rasgos peculiares que les diferenciaban del resto y que, a su vez, les configuraban como un grupo homogéneo. Ello condujo a la descripción inicial de la HOP a partir de características clínicas, radiológicas y anatomopatológicas de 5 casos1. La comunicación de nuevos casos4-10 ha permitido ampliar la información sobre el mecanismo íntimo de esta forma de osificación ectópica, y conocer más a fondo el origen del proceso11. Aunque se trata de un trastorno muy poco frecuente, creemos que puede tener interés presentar por primera vez esta entidad en nuestro medio, máxime en un ámbito especializado como el que atiende nuestra revista. Conviene recordar, además, que algunas de estas enfermedades, de aparición rara, pero con rasgos patológicos bien definidos, se pueden considerar como experimentos naturales cuya investigación permite extraer conclusiones que pueden ser aplicadas con éxito a resolver problemas comunes. Después de describir el caso que hemos tenido la oportunidad de estudiar en colaboración con el profesor Kaplan, realizaremos un análisis comparativo de las características diferenciales de la HOP respecto a otras causas de osificación heterotópica, en particular con la FOP y la osteodistrofia hereditaria de Albright (OHA), y concluiremos con un comentario sobre las enseñanzas que podemos obtener del proceso que nos ocupa.
Descripción del caso
J.G.M. nació en 1966, en Las Palmas de Gran Canaria, producto de un parto prematuro (al octavo mes de gestación), resuelto por cesárea debido a la presencia de una placenta previa con hemorragia aguda. No había ningún antecedente conocido de enfermedad ósea hereditaria en la familia. Sus padres no tenían relación de parentesco entre sí y, en el momento del parto, tenían 18 (la madre) y 21 años de edad, respectivamente. J.G.M. era el mayor de tres hermanos. Su hermano, diagnosticado de síndrome de Down, murió al poco de nacer. Su hermana, aunque con talla y peso bajos al nacer, por lo demás era normal.
J.G.M. pesó al nacer 2,8 kg (percentil 25). En ese momento, no se apreció acortamiento del primer dedo de los pies ni ninguna otra malformación esquelética. Unos meses después apareció una lesión cutánea de aspecto eritematoso localizada sobre la región del quinto metacarpiano de la mano izquierda y se observó una placa indurada en el antebrazo del mismo lado. Durante el año siguiente, sin que mediaran traumatismos o algún otro factor que lo precipitara, desarrolló nuevas máculas eritematosas de pequeño tamaño localizadas en el párpado y en la piel correspondiente a la región occipital. En poco tiempo, ambas se transformaron en placas firmes, no dolorosas ni inflamatorias. Se inició un tratamiento con corticoides por vía oral, que tuvo que ser suspendido a los pocos meses debido a la ausencia de resultados beneficiosos y a la presencia de efectos secundarios importantes. A los 2 años de edad fue trasladado a una unidad pediátrica especializada donde se confirmó la presencia de placas subcutáneas osificadas en la mano y el antebrazo izquierdos, así como en la zona occipital posterior. Además, se observó la presencia de nuevas lesiones en forma de placas o nódulos, de idénticas características a las previas, en la mano derecha, las muñecas, la clavícula izquierda y el talón izquierdo. Se realizó un estudio bioquímico completo, que incluía la determinación de calcio y fósforo en sangre y orina, y fosfatasa alcalina total, todo lo cual resultó normal para su edad. Se apreció un desarrollo psicomotor normal, salvo un leve retraso en el lenguaje. Se realizó una biopsia de dos de las placas subcutáneas mencionadas. En ambas preparaciones se observó una epidermis normal y la formación de hueso heterotópico, sobre todo alrededor de los vasos, los cuales aparecían congestivos. En una de ellas, la formación de hueso se localizaba en la dermis reticular mientras que en la otra se encontraba en la hipodermis. Era un tejido óseo de tipo inmaduro (fibrilar) y con un patrón de osificación de predominio intramembranoso. Con el diagnóstico de calcinosis cutis se prescribió un tratamiento con suplementos minerales. Un año más tarde, las lesiones preexistentes no habían progresado ni habían aparecido nuevos elementos, pero en la siguiente revisión, a los 5 años de edad, las placas del brazo izquierdo presentaban claros signos de reactivación. Además, apareció sobre la zona de la axila izquierda un nódulo de consistencia firme, que también se osificó rápidamente. Con excepción del nódulo situado en la planta del talón izquierdo, que por resultarle molesto al caminar hubo de ser resecado, ninguna de las lesiones descritas le ocasionaba dolor. Sin embargo, experimentó una limitación progresiva de la movilidad articular en las regiones comprometidas, en particular en la muñeca y en el hombro izquierdos. A la edad de 7 años presentaba placas de osificadas en ambas manos (más extensas en la izquierda), el antebrazo izquierdo, la clavícula, el hombro y la axila del mismo lado (extendiéndose hacia la pared torácica adyacente), la región laterocervical izquierda y la zona occipital posterior. No se registró ningún cambio notable hasta los 12 años de edad, en que, por la limitación funcional que le ocasionaba, se decidió extraer la placa de osificación localizada sobre el antebrazo izquierdo y, asimismo, se realizó la resección de la cabeza del radio adyacente. Sin embargo, a los pocos meses dicha lesión recurrió parcialmente, quedando el codo en anquilosis parcial. Además, se produjo una cicatrización queloidea en el lugar de la intervención. A los 18 años de edad desarrolló nuevas placas en el músculo recto anterior del abdomen que rápidamente se endurecieron, ocasionándole una importante disminución de la movilidad, por lo que fueron resecadas, esta vez con buenos resultados. En este período el diagnóstico inicial se sustituyó por el de miositis osificante. Poco después se le practicó una oseotomía de Darrach sobre la muñeca izquierda. Durante el acto quirúrgico presentó una parada cardíaca de la que se recuperó sin secuelas.
A los 19 años de edad, tras rebasar la edad pediátrica, fue remitido por primera vez a nuestro servicio. Las múltiples placas osificadas desarrolladas hasta entonces habían permanecido estables en los últimos años. Presentaba una intensa disfonía que se atribuyó a una parálisis de las cuerdas vocales. En la exploración física se apreciaba una alopecia focal y una discreta distrofia ungueal. La movilidad cervical se encontraba muy limitada, al igual que todos los movimientos del hombro izquierdo. El codo izquierdo estaba anquilosado en extensión y la movilidad de las muñecas era muy limitada. Se confirmaron las placas de osificacón ectópica, ninguna de las cuales presentaba signos de actividad, en las localizaciones ya referidas. No se observaron anomalías en las rodillas ni en las caderas. Tampoco se apreció un acortamiento de los dedos ni otras malformaciones esqueléticas. El balance metabólico óseo era normal, al igual que los marcadores de formación (fosfatasa alcalina en suero) y resorción (eliminación total de hidroxiprolina en orina de 24 h). En el examen radiológico se apreciaban masas calcificadas con patrón fibrilar (sin formación de hueso maduro) en el hombro, el codo, el antebrazo y la mano izquierdos (figs. 1-3). Se apreciaba también la formación de un bloque posterior en los cuerpos vertebrales C2 a C5, con fusión de las articulaciones interapofisarias (fig. 4). No se apreciaban alteraciones en el tamaño o la forma de los huesos metatarsianos y metacarpianos. Con el diagnóstico de fibrodisplasia ósea progresiva se le recomendó evitar traumatismos y toda intervención quirúrgica que no fuera estrictamente indispensable, y se indicó tratamiento con etidronato (400 mg/día) que se mantuvo durante 6 meses sin resultados aparentes. Desde ese momento y hasta hace 2 años, en que tras revisar el caso se consideró que podría tratarse de una HOP, el paciente no volvió a la consulta. Durante estos años, en los que tuvo dos hijos sanos, la enfermedad permaneció prácticamente estacionaria permitiéndole realizar una vida activa e incluso practicar deporte, con una disminución de la movilidad en la región cervical y la extremidad superior izquierda. La última valoración analítica, realizada cuando el paciente contaba 35 años de edad, que incluyó los más recientes marcadores del remodelado óseo, continúa siendo normal. Se enviaron muestras apropiadas de sangre al laboratorio del Departamento de Medicina Ortopédica Molecular de la Universidad de Pensilvania (Dra. Eileen Shore), donde se realizó un estudio completo de la región del gen GNAS1, no detectándose ninguna mutación en dicha área. En la exploración radiológica se constató la ausencia de modificaciones en las lesiones conocidas, observándose además dos lesiones calcificadas de muy pequeño tamaño adyacentes a la cara interna de la rodilla y en el pie. Se realizó, además, una gammagrafía ósea de cuerpo completo con 99mtecnecio-EHDP, que demostró depósitos patológicos del trazador, de intensidad variable, en las zonas donde existían placas calcificadas (fig. 5).
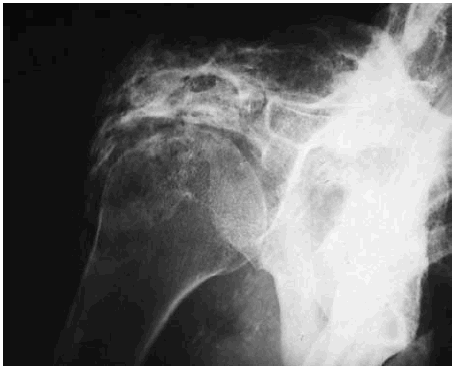
Figura 1. Calcificación ectópica periarticular de aspecto fibrilar en la región del hombro.
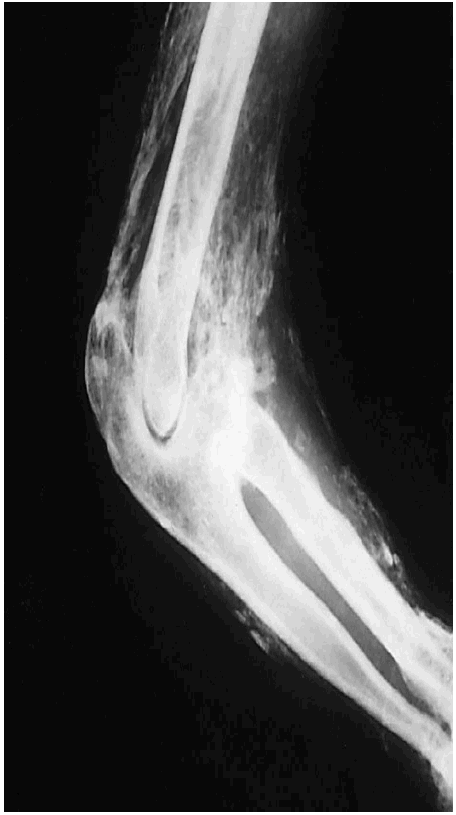
Figura 2. Extensa calcificación ectópica fibrilar que sigue los haces musculares del brazo, el codo y el antebrazo.
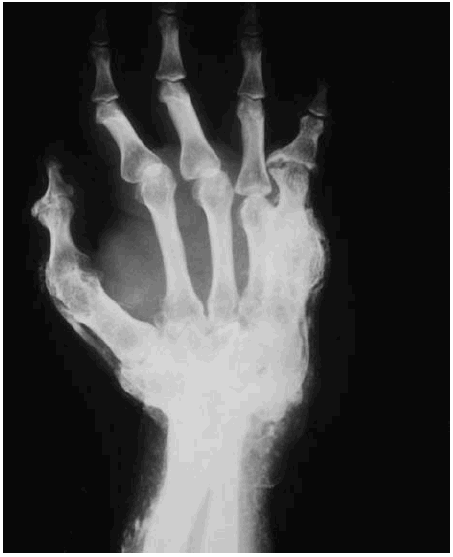
Figura 3. Calcificación subcutánea y muscular en el antebrazo y la mano, sobre todo en el cuarto y el quinto dedos, con clinodactilia y notable displasia ósea que ocasiona cambios degenerativos secundarios.
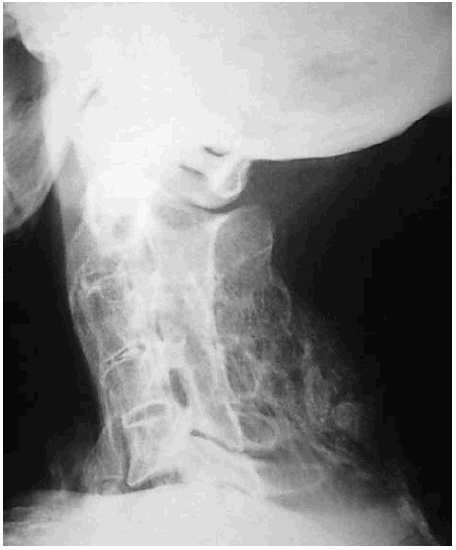
Figura 4. Bloque cervical por fusión de las articulaciones interapofisarias (C2 a C5) y de otras estructuras del segmento vertebral posterior.
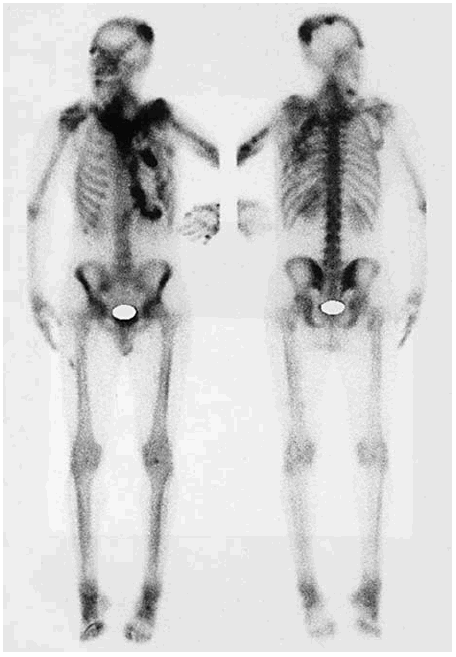
Figura 5. Depósitos patológicos del radiotrazador (99mtecnecio-EHDP) en el área occipital, el hombro izquierdo, la unión cérvico-esternal y la región costal anterior izquierda.
Calcificación heterotópica de predomino cutáneo y muscular
La presencia de calcificación y osificación fuera del tejido óseo (hueso ectópico) se presenta en un amplio número de enfermedades muy diversas (tabla 1). Algunas, como la calcificación de estructuras paraespinales de la espondilitis anquilosante, la calcificación distrófica de la piel en la esclerodermia, o la mal llamada «calcificación metastásica» del hiperparatiroidismo, nos resultan muy familiares a los reumatólogos. En el amplio grupo debido a la formación, por células mesenquimatosas con diferenciación osteoblástica, de tejido óseo en lugares inadecuados (hueso heterotópico), la afección del músculo y de la piel es muy relevante. Además de en ciertos procesos adquiridos (enfermedades neurológicas, traumatismos...), este tipo de osificación anómala ocurre en un grupo de enfermedades congénitas cuyos mecanismos etiopatogénicos han sido, en parte, descifrados: la FOP, la HOP y la OHA. Las principales diferencias entre estas entidades se resumen en la tabla 2. En la FOP y en la HOP la osificación se extiende a planos musculares profundos, mientras que en la OHA se limita a la piel y al tejido conjuntivo superficial11. Por otra parte, los pacientes con FOP o HOP no presentan los rasgos somáticos (baja estatura, obesidad, cara redonda y acortamiento de los metacarpianos cuarto y quinto) ni las anomalías endocrinas que caracterizan a la OHA12,13. La alteración molecular de la OHA es una mutación que inhibe el gen GNSA1 (cromosoma 20q13)14-19, el cual codifica la subunidad alfa de la proteína G (Gsα). Este defecto genético ocasiona una disminución en la actividad de la adenilciclasa y una alteración en la señal de transducción mediada por el AMPc, y puede producir dos fenotipos, según exista resistencia a la PTH (seudohipoparatiroidismo tipo Ia) o no (seudo-seudohipoparatiroidismo)20,21.

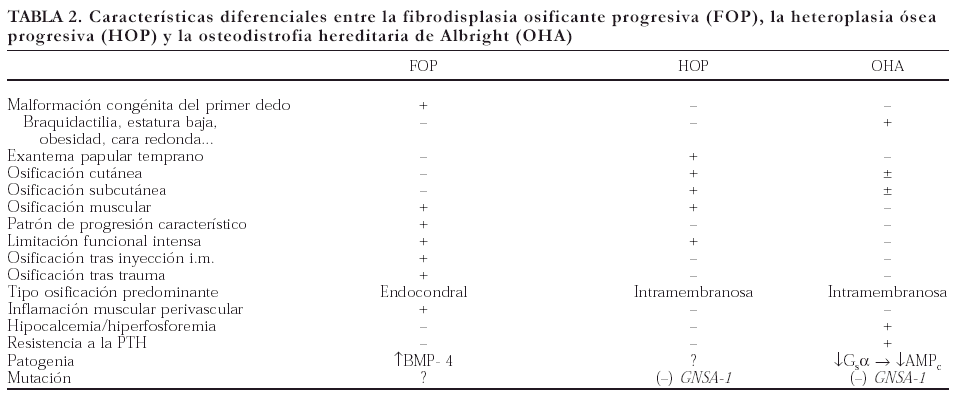
Las diferencias entre la FOP y la HOP son algo más sutiles; de hecho, hasta hace poco, ambas enfermedades se confundían, aunque ahora sabemos que son tan nítidas que pueden considerarse entidades claramente diferenciadas11. Se ha indicado que la HOP se puede distinguir de la FOP por la presencia de calcificaciones cutáneas, la ausencia de malformaciones esqueléticas congénitas (en particular en el modelado del primer dedo del pie), la distribución asimétrica de las placas calcificadas, las diferencias en la evolución topográfica de las placas de osificación y el predominio de osificación intramembranosa (en lugar de endocondral)1. Al igual que en la FOP, las determinaciones analíticas del metabolismo calcio-fósforo suelen ser normales en la HOP, aunque se puede observar un aumento transitorio en las cifras de fosfatasa alcalina (en las fases de osteogénesis más activa), así como una elevación moderada de la LDH y la CPK ocasionada por la destrucción de fibras musculares debida al depósito de hueso en ese tejido8,9. La estabilización del proceso al final de la adolescencia y la ausencia de placas de hueso lamelar maduro en los planos musculares y las zonas periarticulares, que ocasionan el fallecimiento temprano de la mayoría de los casos de FOP (generalmente por infecciones respiratorias asociadas a calcificación de músculos intercostales), confieren a la HOP un pronóstico más favorable11. Además, a diferencia del invariable y exagerado recrudecimiento que acompaña a cualquier intento de resección en las placas o ante el más mínimo traumatismo (incluso tras una simple inyección intramuscular) en la FOP, en la HOP esta tendencia es menos acentuada e incluso, como ocurrió en nuestro paciente, algunas placas aisladas pueden ser eliminadas con éxito. Sin embargo, dada la escasa experiencia aún disponible, también en la HOP se aconseja evitar cualquier procedimiento quirúrgico que no sea estrictamente indispensable, y el pronóstico de la enfermedad a largo plazo es todavía incierto. Finalmente, el patrón de transmisión, tanto en la HOP como en la FOP y en la OHA, es de tipo autosómico dominante, por lo que no cabe establecer diferencias en este sentido. Considerando las características diferenciales mencionadas, los rasgos del paciente que hemos comentado (J.G.M.) que permiten descartar la presencia de una FOP y que apoyan la existencia de una HOP son los siguientes:
1. Ausencia de malformación congénita del primer dedo.
2. Erupción eritematosa papular cutánea al comienzo de la enfermedad.
3. Calcificación cutánea y subcutánea.
4. Ausencia de placas musculares calcificadas después de traumatismos o inyecciones.
5. La evolución del patrón topográfico de las placas calcificadas: de axial a apendicular, de craneal a caudal y de proximal a distal, al revés de lo que es habitual en la FOP. Más aún, la mayoría de las lesiones aparece en un solo lado del cuerpo (en este caso el izquierdo), una característica cuya especificidad ha sido señalada por otros autores5.
6. Las características radiológicas de la lesión básica: masas calcificadas con «patrón fibrilar» (figs. 1-4) en lugar de las grandes cintas o puentes de hueso maduro características de la FOP.
¿Qué podemos aprender de un proceso de aparición tan infrecuente?
La HOP se puede concebir como una anomalía en el desarrollo y la diferenciación del mesénquima, caracterizada por la aparición muy temprana de placas calcificadas en la dermis, con desarrollo progresivo, durante el resto de la infancia y la juventud, de áreas de osificación heterotópica en estructuras de tejido conjuntivo más profundas y en el propio músculo estriado. Kaplan et al, en su artículo de revisión publicado en noviembre de 200011, señalaban que hasta ese momento habían sido publicados 16 casos de HOP (13 mujeres y 3 varones) e indicaban tener conocimiento de 13 casos adicionales (4 mujeres y 9 varones). Casi todos los casos parecen ser esporádicos aunque se ha descrito algunas familias en las que uno o varios parientes cercanos del propositus presentaban calcificaciones cutáneas1. Aunque la ausencia de familias amplias impide la identificación de genes de susceptibilidad mediante análisis de ligamiento y clonación posicional, se han realizado estudios de análisis de mutación sobre genes candidatos. Es este sentido, la presencia de una mutación inhibidora en el gen GNSA1 en tres casos de calcinosis cutis22,23, una entidad que ahora se considera una variante clínica de la HOP, preludió el hallazgo, recién publicado, de esta misma mutación en la propia HOP24. Ello sugiere que las mutaciones que inhiben este gen pueden ocasionar un espectro de manifestaciones que abarca desde la calcificación cutánea o subcutánea aislada (calcinosis cutis y OHA) hasta formas de calcificación y osificación más profundas (HOP), asociadas o no con malformaciones esqueléticas (OHA, ¿HOP?), con o sin alteraciones hormonales (OHA). La base molecular que sustenta tal serie de alteraciones resulta congruente con los datos experimentales disponibles. El gen GNSA1 codifica la subunidad alfa de la proteína G (Gsα). Se sabe que la PTH y el péptido relacionado con esta hormona (PTHrP) utilizan un receptor de membrana común asociado a la proteína Gsα25,26. Sin embargo, el papel fisiológico de este receptor va más allá de la mera regulación de la homeostasis fosfocálcica ya que, además, es clave en el desarrollo embrionario de hueso y cartílago26,27 e interviene en la maduración y la actividad de los osteoblastos26,28. Una mutación que inhiba el gen GNSA1 podría interrumpir la señal para la transducción del PTHrP en el embrión y, presumiblemente, ocasionar las alteraciones (talla baja, braquidactilia y calcificaciones subcutáneas) características de la OHA. Sin embargo, se desconoce el mecanismo molecular por el que esta mutación puede dar lugar a la osificación progresiva de los tejidos conjuntivos profundos y el músculo. La hipótesis más obvia es que se produciría una alteración en la señal mediada por AMPc en «células tronco» de origen mesenquimatoso. En este sentido, es muy interesante la constatación de que la mutación activadora del gen GNSA1, que origina el síndrome de McCune-Albright (¡curiosa coincidencia que el genial Fuller Albright realizara la descripción clínica de dos procesos distintos en los que, 50 años más tarde, se han demostrado mutaciones de signo opuesto en el mismo gen!), dé lugar a un freno al desarrollo de la osteogénesis en los huesos afectados (displasia fibrosa). Este hecho refuerza la idea de que el gen GNSA1 codifica la síntesis de un factor regulador de la osteogénesis que tiene efectos negativos sobre desarrollo de las células de estirpe osteoblástica. El estudio de nuevos casos de HOP puede permitir el descubrimiento de este factor y el esclarecimiento de los mecanismos moleculares que regulan la expresión génica asociada a la transformación de células precursoras de la piel, grasa y músculo en células óseas maduras. Así, la HOP puede contribuir a una mejor comprensión de enfermedades, mucho más frecuentes, en las que existe un trastorno de la osteogénesis, y a la consecución de un tratamiento eficaz.
Agradecimientos
Al Profesor Prederick Kaplan por sus expertos comentarios, de inestimable ayuda en la valoración de este paciente, y por la realización de los análisis genéticos. A los laboratorios PHARMACIA, cuya colaboración nos ha permitido completar el estudio.