


Sr. Director: La enfermedad de Kikuchi (EK) o linfadenitis necrosante histiocítica es una entidad poco común, caracterizada por linfadenopatía cervical y/o axilar, fiebre, lesiones cutáneas, leucopenia ocasional y disfunción hepática, cuyo curso clínico suele ser benigno y de duración autolimitada. En 1972, Kikuchi1 y Fujimoto et al2 describieron independientemente las características clínicas e histológicas de la enfermedad, aunque se han recogido observaciones históricas en informes del Centro de Referencia de Patología del Instituto Nacional del Cáncer en Estados Unidos en 19643. La presencia de otras entidades como el lupus eritematoso sistémico (LES) y el síndrome hematofagocítico (SH), que presentan síntomas comunes a la EK, hace difícil, en muchos casos, el diagnóstico definitivo, especialmente cuando se presentan de forma simultánea, lo que se presta a confusión y aumenta el desafío diagnóstico en esos casos. Presentamos un caso con manifestaciones comunes de LES y EK en remisión total después de un año de enfermedad.
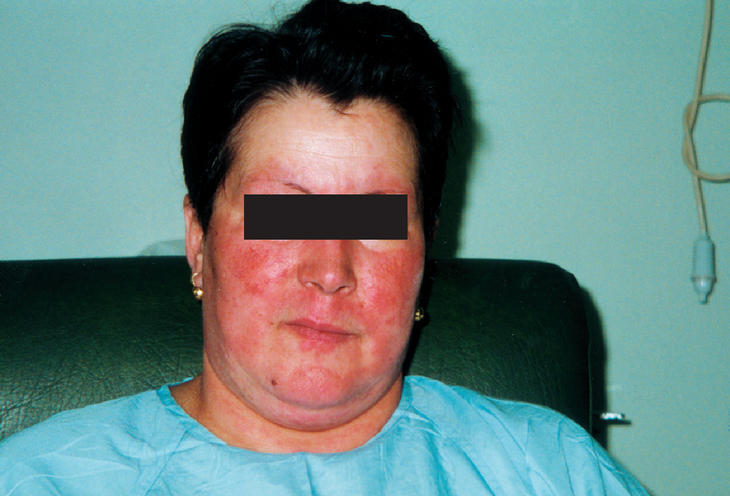
Paciente de 49 años, con manifestaciones clínicas comunes de LES y EK de comienzo agudo y simultáneo. Una biopsia de ganglio evidenció la presencia de hallazgos compatibles con EK. La aparición de manifestaciones clínicas orgánicas mayores precisó de un tratamiento más agresivo, y la evolución clínica fue favorable. Se comentan los hallazgos clínicos, etiopatogénicos y patológicos que caracterizan a dichas enfermedades y su diagnóstico diferencial con el SH. La paciente había sido estudiada en el servicio de dermatología por presentar lesiones cutáneas maculares eritematosas dispersas por la cara, superficie anterior del tórax y extremidades superiores, con prurito ocasional durante los 2 años previos a su actual ingreso. Ingresó por presentar un cuadro de 2 meses de evolución con exantema facial macular eritematoso (fig. 1) en cara, cuello y extremidades inferiores, fiebre, artralgias en hombros, codos, manos, rodillas y pies con signos de sinovitis en muñecas y adenopatías cervicales y axilares indoloras. También presentaba una lesión nodular en la mama derecha. Entre los antecedentes familiares, 2 tías habían fallecido por cáncer de mama. Durante su ingreso se detectaron los siguientes resultados: anemia con hemoglobina, 10 g/dl (normal, 12-16); volumen corpuscular medio y concentración media de hemoglobina corpuscular, normales; leucopenia, 1.700 leucocitos (normal, 4.500-11.000) y ligera trombopenia, 100.000 plaquetas (normal, 130.000-400.000); elevación de las transaminasas GOT, 110 U/l (normal, 10-37); GPT, 48 U/l (normal, 7-40) y gGT, 59 (normal, 10-35); los cultivos de sangre y orina resultaron normales y el Mantoux fue negativo. Una mamografía bilateral mostró un discreto aumento difuso de densidad del tejido mamario derecho con engrosamiento cutáneo y masas adenopáticas de 2-3 cm en la región mamaria derecha y una adenopatía axilar izquierda de 1,5-2,5 cm. La radiografía de tórax y la ecografía abdominal no mostraron alteraciones. La tomografía axial computarizada (TAC) toracoabdominal evidenció adenopatías axilares. Ante la posibilidad de una enfermedad infecciosa, inflamatoria o neoplásica, se realizaron otros estudios. En principio, la paciente fue tratada con amoxicilina-ácido clavulánico 1 g/8 h y imipenem 500 mg/6 h por vía intravenosa (i.v.), y antiinflamatorios no esteroideos.
Figura 1. Exantema facial con localización preferente en área malar y nasal.
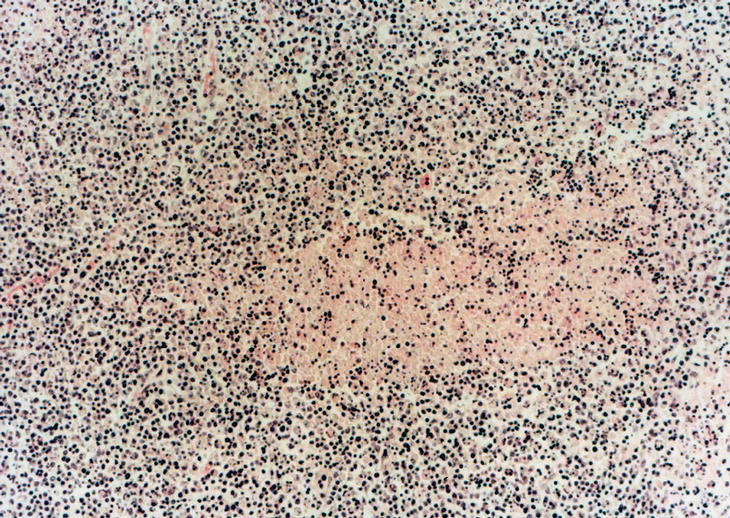
Una punción aspiración del nódulo axilar izquierdo mostró una lesión granulomatosa sin signos de malignidad, con BAAR negativo. Los estudios de serología de lúes, marcadores de hepatitis, virus de la inmunodeficiencia humana (VIH), herpes tipo VI, anticuerpos anticitoplasmáticos (ANCAS) y marcadores tumorales resultaron negativos, con títulos elevados de IgG para parvovirus B19, toxoplasma, virus de Epstein-Barr, citomegalovirus y herpes simple. Los anticuerpos antinucleares (ANA) resultaron positivos a un título de 1/320 y anti-DNA de 1/10 (normal, < 1/10), con una fracción C3 y C4 de complemento bajos (C3, 53 normal, 88-20; C4, 7 normal, 16-47). Se realizó una biopsia de ganglio axilar derecho, y la paciente fue diagnosticada de LES; se trató con prednisona en dosis de 1 mg/ kg/día en el momento de su alta. Al cabo de 15 días acudió a las consultas por presentar cuadro de bradipsiquia y confusión, con inestabilidad en la marcha y desorientación parcial espacial y temporal sin acompañarse de cefalea ni fiebre. La TAC cerebral resultó normal y la resonancia magnética nuclear (RMN) cerebral mostró pequeñas imágenes de hiperintensidad de señal en sustancia blanca periventricular y ganglios de la base inespecíficos. Se realizó una punción lumbar en la que se observó una discreta hipercelularidad con aumento de proteínas, con cultivos BAAR y ADA negativos. Inicialmente se trató con aciclovir y ampicilina de forma empírica, ante la posibilidad de una meningoencefalitis infecciosa. El resultado de la biopsia de ganglio evidenció la existencia de linfadenitis necrosante no supurativa con áreas de necrosis, e infiltrado histiocitario y linfoplasmocitario con áreas de cariorrexis, todo ello compatible con EK (fig. 2).
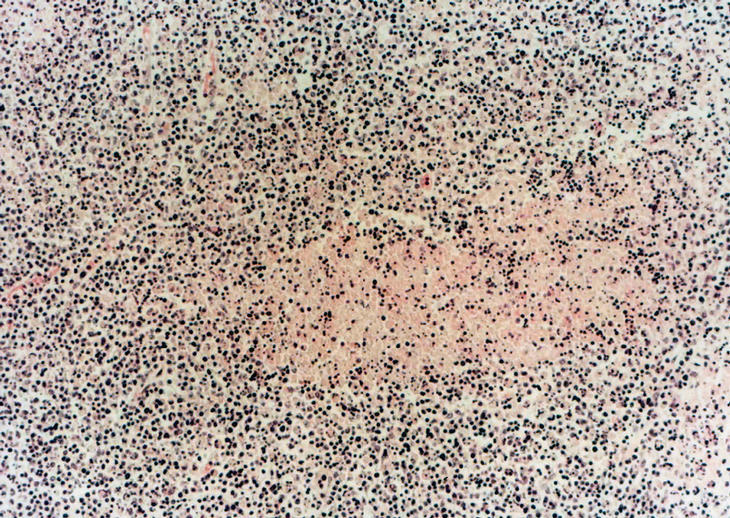
Figura 2. Biopsia de ganglio con áreas de necrosis e infiltrado histiocitario y linfoplasmocitario. Presencia de cariorrexis.
Los síntomas neurológicos fueron interpretados como una manifestación clínica debida al LES en el seno de una vasculitis en la sustancia blanca periventricular y ganglios basales (exantema facial, artralgias, pancitopenia, ANA+, anti-ADN+, hipocomplementemia, cambios en la RMN cerebral); se trató con pulsos mensuales de ciclofosfamida (1g/i.v.) durante 7 meses y pauta descendente de prednisona, y un año después se encontraba en remisión total, asintomática y sin tratamiento, con el título de ANA de 1/80 y anti-ADN negativos con una fracción C3 de complemento de 75. Todas las alteraciones hematológicas y hepáticas se normalizaron. Clínicamente, no se puede descartar de forma definitiva que los síntomas neurológicos no fueran debidos a una consecuencia de la EK (acompañados de pancitopenia y exantema facial).
La linfadenitis necrosante de Kikuchi-Fujimoto (EK) es una entidad poco común, de curso generalmente autolimitado incluso sin tratamiento (aunque en la bibliografía se han descrito algunos casos fatales)4 y que precisa confirmación histológica para su diagnóstico. La enfermedad presenta una marcada predilección por las mujeres (80% de los casos publicados), y aunque ha sido descrita en un amplio rango de edad (11-75 años), la mayoría tienen menos de 30 años.
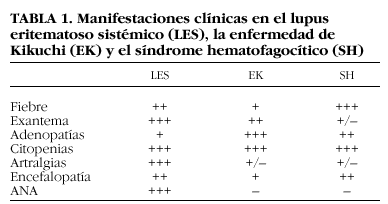
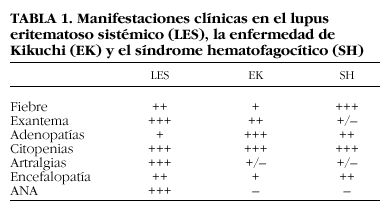
La EK tiene características clínicas comunes con el LES y el SH, con casos clínicos descritos en la bibliografía de asociación entre dichas entidades5-7. Hay autores que incluso piensan que pueden tratarse de distintas fases de una misma enfermedad8 (tabla 1).
En nuestra paciente se realizó el diagnóstico diferencial con la tuberculosis (TB) ganglionar, el linfoma y el cáncer de mama, entre otros. El cáncer de mama fue la primera posibilidad diagnóstica ante la aparición de un nódulo en una mama y los antecedentes familiares. Por otra parte, la presencia de manifestaciones lúpicas nos indujo a considerar las características clínicas y biológicas que caracterizan al LES y a la EK; en nuestro caso cumplía los criterios clínicos de LES (con ANA y anti-ADN+) y EK (biopsia ganglionar). Otra enfermedad a tener en cuenta en el diagnóstico diferencial era el SH, que se caracteriza por presentar manifestaciones clínicas comunes a las anteriores; sin embargo, la ausencia de manifestaciones más graves y de hematofagocitosis relegaban esta enfermedad a un segundo plano.
En la EK consideramos que algún agente ambiental desconocido, posiblemente infeccioso, desencadena una respuesta inmune, lo que provoca una activación de los linfocitos T con secrección de citocinas, que dan lugar a las manifestaciones clínicas conocidas. La presencia de hiperplasia ganglionar como respuesta a una agresión desconocida indicaría la activación de una población linfocitaria en los órganos linfoides secundarios. La falta de respuesta por parte de las células ubicadas en los órganos del sistema reticuloendotelial para la eliminación de los agentes inflamatorios hace que se produzca una respuesta exagerada en esos órganos linfoides con hiperplasia de éstos, que se manifiesta por la presencia de adenomegalias y hepatoesplenomegalia.
La asociación más común con las enfermedades del tejido conectivo y las neoplasias hace pensar en una respuesta aberrante del sistema inmunitarios, incapaz de eliminar los antígenos y depósitos apoptóticos que se producen. De hecho, aunque la causa no está clara, se ha sugerido que la apoptosis es importante en la patogénesis de la EK. Aunque la población CD4 también está representada, se ha sugerido que los linfocitos CD8 serían células efectoras apoptóticas así como células diana, mientras que los histiocitos acentuarían la apoptosis en la EK9. De hecho, esos restos apoptóticos actuarían a su vez como autoantígenos que perpetúan y agravan la enfermedad, como lo prueba el hecho de encontrar fragmentos de ADN en las áreas de los ganglios linfáticos afectados, con células positivas para anticuerpos anti-FAS10.
La asociación de LES y EK en forma de presentación aguda con fiebre y síntomas generales, apunta a que sea un agente infeccioso el factor desencadenante en el seno de una alteración inmunitaria aberrante incapaz de eliminar los productos resultantes de la apoptosis11. De hecho, una regulación desordenada de la apoptosis y del aclaramiento de los productos apoptóticos han sido implicados en la patogénesis del LES12,13.
Una deficiencia en las fracciones C1 y C1q del complemento se ha relacionado con el aumento de riesgo de LES14, ese hallazgo produciría una respuesta humoral aberrante con activación de los linfocitos B y alteración en el aclaramiento de las células apoptóticas y complejos inmunitarios. Cualquier agente inductor de apoptosis podría provocar alteraciones en la eliminación de los restos apoptóticos, como sucede en el LES y, por ello, cabría preguntarse si determinados tratamientos (coxib), en los que se ha comprobado cierta inducción de apoptosis, podrían ser contraproducentes en el tratamiento de determinadas afecciones en las que una apoptosis alterada favorecería la presentación de enfermedad15.
Otro dato a tener en cuenta en la EK, es que la enfermedad es más severa en pacientes con afección de la piel. Parece ser que las células que infiltran las lesiones cutáneas están compuestas por los mismos componentes que las lesiones en los ganglios linfáticos. Takakuwa et al16 demostraron que las proteínas que regulan la apoptosis, bcl-2, bax, c-myc y p53, no estaban involucradas en el desarrollo de células necrosantes, pero que la perforina, una proteína específica de células killer esencial que provoca apoptosis en las células diana, estaba expresada de forma abundante en las células infiltrantes, las cuales parecían ser células T citotóxicas.
La situación inmunitaria aberrante que se produce en el lupus y su asociación con la EK hacen pensar en la posibilidad de que determinados autoantígenos, ya sean por mecanismos proapoptóticos o por dificultades en su eliminación, tengan un papel preponderante en la enfermedad y pueden agravar la enfermedad de base.
El SH17 es un proceso histiocítico reactivo, frecuentemente asociado a infección o malignidad y suele ser letal en el 50% de los casos, en muchos casos acompaña a una coagulopatía con elevación de los productos derivados de la fibrina. Una hiperplasia de células de Kupffer con hemofagocitosis es característica de la enfermedad, con aumento del número de histiocitos dentro de los sinusoides hepáticos y hemofagocitosis18. El exámen de médula ósea es diagnóstico, y se observa una marcada hiperplasia histiocitaria con hematofagocitosis y concentraciones altas de IL-1ß e IL-6. La buena respuesta que en muchos casos de SH sigue al tratamiento con ciclosporina A, hace pensar en un desorden de células T en relación con la enfermedad, que además se demuestra por la disminución del receptor de la IL-2 en esos casos19. Además, como consecuencia de la disminución de la función celular con el tratamiento también se inhibe la secreción de interferon-Y, que ha demostrado tener un papel fundamental en la activación del macrófago, todo ello clave en la patogénesis del SH. Algunos estudios sugieren que la IL-18 es importante en la patogénesis del SH, particularmente a través de la inducción de los linfocitos TH1, y además se observa una buena correlación entre las concentraciones de IL-18, interferón-* y el ligando del Fas soluble; así, la medición sérica de IL-18 puede ser de ayuda para el diagnóstico y detección de la actividad de la enfermedad20. Por otra parte, en algunos estudios se han detectado concentraciones aumentadas en la expresión de las moléculas de clase I y II del MHC y de las moléculas de adhesión LFA-1, LFA-3 y ICAM-1 en los macrófagos esplénicos21.
Por consiguiente, la EK y el SH son enfermedades con características clínicas comunes que pueden asociarse entre sí y también con el LES, y pueden representar un importante desafío diagnóstico en determinados casos, llegando en ocasiones a superponerse a la enfermedad de base. En muchos casos, el pronóstico depende del diagnóstico temprano y del tratamiento aplicado y, por ello, es fundamental su conocimiento en el contexto o no de una enfermedad infecciosa, autoinmune o neoplásica. La decisión de tratar con una terapia más agresiva dependerá de la presencia de una entidad u otra y de las manifestaciones clínicas en cada caso. La aparición de nuevas terapias biológicas y la presencia de concentraciones altas de factor de necrosis tumoral alfa (TNF-*) e INF-* en el SH hace pensar en la terapia con anti-TNF como una alternativa nueva en el tratamiento de dicha entidad.