


Introducción
El síndrome antisintetasa se caracteriza por polimiositis (PM) inflamatoria, afectación pulmonar intersticial, artritis, fiebre, «manos de mecánico», afectación del estado general, anticuerpos frente a ribonucleoproteínas citoplasmáticas implicadas en el proceso de translación (anticuerpos antisintetasa) y respuesta variable al tratamiento. Puede acompañarse de fenómeno de Raynaud, que suele estar presente al inicio de los síntomas musculares. Presentamos un paciente con un síndrome antisintetasa precedido de fenómeno de Raynaud unilateral severo 3 años antes. Dicho cuadro se interpretó como ocupacional y fue tratado mediante simpatectomía torácica, el paciente permaneció paucisintomático hasta el desarrollo de la sintomatología articular y muscular. Al inicio del cuadro no se llevaron a cabo procedimientos destinados a descartar otras causas de fenómeno de Raynaud secundario.
Caso clínico
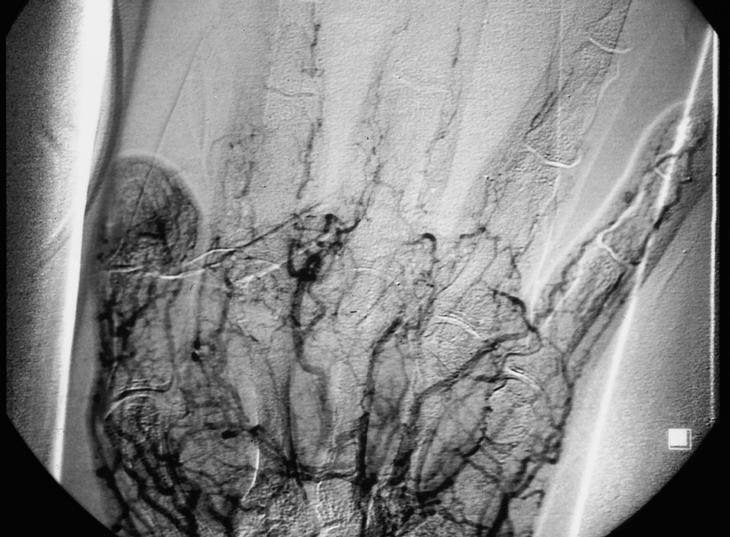
Varón de 55 años, tornero de profesión, con antecedentes de psoriasis de 30 años de evolución, y fractura traumática de L-4. En 1998 fue estudiado en nuestro centro por cuadro de 3 meses de evolución de fenómeno de Raynaud severo, con isquemia en segundo, tercero y cuarto dedos de la mano derecha. La arteriografía evidenció oclusión de arterias interdigitales de los 3 dedos medios de la mano derecha (fig. 1). El hallazgo se atribuyó a síndrome ocupacional, y 2 meses después se trató mediante simpatectomía torácica. En la radiografía de tórax realizada en aquél momento, no se evidenciaron alteraciones parenquimatosas. Posteriormente, siguió controles en cirugía vascular, y persistía un fenómeno de Raynaud leve, especialmente en invierno. El paciente continuó con su actividad laboral habitual.
Figura 1. Arteriografía de miembro superior derecho. Oclusión de arterias interdigitales del segundo, tercero y cuarto dedos, con ausencia total de relleno y mal relleno del arco palmar.
A finales del año 2001 fue remitido a nuestra unidad por clínica de un mes de evolución de poliartralgias de ritmo inflamatorio en muñecas e interfalángicas proximales, junto con rigidez matutina de 1 h. En la exploración realizada en el momento de la consulta se objetivó leve derrame articular en rodilla derecha y sinovitis en segunda interfalángica proximal izquierda, el resto de la exploración fue anodina y se solicitaron estudios analíticos. Estando pendiente de revisión, ingresó por disnea progresiva de 4 semanas de evolución junto con fiebre, expectoración oscura, cefalea, aumento de la intensidad de las artralgias y pérdida de fuerza en extremidades.
A su ingreso presentaba temperatura de 37,4 °C, tensión arterial de 123/75 mmHg, saturación de 02 del 83% y frecuencia cardíaca de 110 lpm. En la exploración física destacaba la presencia de crepitantes bibasales de predominio izquierdo y la pérdida de fuerza proximal en extremidades superiores e inferiores.
El hemograma y la bioquímica de rutina fueron normales, a excepción de: GOT, 395 U/l; GPT, 355 U/l, y CPK, 6.957 U/l. La proteína C-reactiva estaba elevada (77 mg/l). El factor reumatoide fue positivo, 299 UI/ml. Los anticuerpos antinucleares (ANA) fueron negativos, con positividad para los anticuerpos anti-histidyl-ARNt sintetasa (anti-Jo-1).
La radiografía de tórax puso de manifiesto una afectación mixta alveolo-intersticial bilateral de predominio en campos inferiores. La tomografía axial computarizada (TAC) de alta resolución, evidenció un infiltrado en vidrio deslustrado, de predominio en lóbulos inferiores junto con parcheados de aumento de densidad periférica. Las pruebas de función respiratoria mostraron alteración ventilatoria restrictiva severa con acusado descenso de la difusión de CO.
Un electromiograma evidenció potenciales de unidad motora de características miopáticas y fibrilaciones y ondas positivas evidentes-profusas en músculos explorados. La biopsia del músculo deltoides mostró una arquitectura conservada con necrosis de fibras musculares aisladas o en pequeños grupos, predominantemente perimisiales, infiltrado por linfocitos y macrófagos con fenómenos de regeneración aislados.
A la vista de la clínica, la analítica, las pruebas de imagen y la electromiografía, el paciente fue diagnosticado de síndrome antisintetasa con afectación muscular y pulmonar. Se inició tratamiento con bolos de 1 g de metilprednisolona/día durante 3 días, seguidos de metilprednisolona a dosis de 1 mg/kg/día y un primer bolo de ciclofosfamida intravenoso.
A pesar de ello, la situación respiratoria empeoró, por lo que se trasladó a la unidad de cuidados intensivos (UCI), y precisó ventilación mecánica no invasiva. Los hemocultivos y cultivos habituales y específicos de esputo fueron negativos. En el lavado broncoalveolar se aisló Pseudomonas aeruginosa, por lo que se inició tratamiento con ciprofloxacino y gentamicina sin que la situación respiratoria mejorara, por lo que se añadieron ciclosporina oral y gammaglobulinas intravenosas (1 g/kg/día durante 2 días) con mejoría.
Se dio de alta con corticoides en pauta descendente, ciclosporina oral (3 mg/kg/día), y bolos mensuales de ciclofosfamida (600 mg/m2).
El paciente ha recuperando progresivamente fuerza muscular, y mantiene las enzimas musculares normales. Persiste la disnea de grandes esfuerzos con disminución de la difusión de CO en las pruebas de función respiratoria y walking-test positivo. En la última TAC realizada (octubre 2002) no se evidenciaron datos de afectación parenquimatosa pulmonar. En la actualidad no refiere fenómeno de Raynaud.
Discusión
El síndrome antisintetasa se caracteriza clínicamente por polimiositis inflamatoria, afectación intersticial pulmonar, artritis, fiebre, manos de mecánico y, analíticamente, por anticuerpos frente a enzimas citoplasmáticas conocidas como aminoacyl-ARNt sintetasas. La etiología y la patogénesis son desconocidas, se han implicado agentes infecciosos, químicos, drogas y mecanismos autoinmunes. Se caracteriza también por una respuesta variable al tratamiento1.
La afectación pulmonar intersticial está presente entre el 5 y el 40% de pacientes con polimiositis/dermatomiositis (PM/DM), y no hay diferencias significativas entre PM o DM2. La presentación clínica puede ser aguda o subaguda, crónica o asintomática y puede preceder, acompañar o suceder a la afectación muscular. La determinación de anticuerpos anti-ARNt sintetasa puede ser útil para predecir el comienzo de miositis o de afectación intersticial pulmonar2. Se ha sugerido que concentraciones elevadas de GOT y ferritina, la presencia de anticuerpos anti-Jo-1 y microangiopatía característica pueden tener valor predictivo y aumentan la necesidad de buscar afectación intersticial pulmonar en los pacientes con PM/DM3. Una glucoproteína de alto peso molecular (KL6) presente en neumocitos alveolares tipo II y células bronquiales epiteliales, puede ser un marcador útil para evaluar la actividad de la afectación pulmonar2,4.
Los anticuerpos antiaminoacyl-ARNt sintetasa se encuentran en el 25-35% de los pacientes con PM/DM, y el más común es el antihistidyl-ARNt (Jo-1) que representa el 20-30% de los casos. Otros anticuerpos son los anti-alanyl'-(PL-12), anti-treonyl-(PL7), antiisoleucyl-(OJ) o antiglicyl-(EJ) ARNt sintetasa2. Bernstein et al, encuentran anticuerpos anti-Jo-1 en el 68% de los pacientes que presentan polimiositis y afectación pulmonar intersticial5.
Puede acompañarse de síntomas constitucionales, fiebre y fenómeno de Raynaud. En un estudio en pacientes con PM, el fenómeno de Raynaud estaba presente en el 62% de los enfermos con síndrome antisintetasa, frente al 30% de los pacientes con PM asociada a anticuerpos anti-Mi-2 y al 26% de los pacientes con PM no asociada a anticuerpos específicos1. Este porcentaje se eleva al 88% en la serie de Bernstein et al5. Se han descrito casos de necrosis digital en pacientes con síndrome antisintetasa6. El fenómeno de Raynaud tiene una alta prevalencia en la población general, y se debe sospechar como secundario en caso de inicio después de los 30 años, cuando los episodios sean intensos, dolorosos, asimétricos o asociados con lesiones isquémicas de la piel y cuando haya datos clínicos de conectivopatía, alteraciones del lecho capilar por capilaroscopia o positividad para autoanticuerpos específicos7.
El pilar del tratamiento de las miopatías inflamatorias idiopáticas continúan siendo los corticoides8, si bien en muchos casos, especialmente en los síndromes antisintetasa, es necesario añadir inmunosupresores como la ciclofosfamida9, ciclosporina10, tacrolimus11 que pueden ser especialmente útiles en la afectación intersticial pulmonar. La fludarabina12 y las inmunoglobulinas intravenosas13 se han utilizado en casos refractarios.
Nuestro paciente presentó un fenómeno de Raynaud severo, con trombosis arterial palmar y digital de la mano derecha demostrada angiográficamente 3 años antes del diagnóstico de PM. El paciente realizaba un trabajo manual con traumatismo en mano derecha (golpes repetidos 50-60 veces al día sobre una palanca), similar al síndrome del martillo hipotenar, pero no utilizaba martillos neumáticos u otros instrumentos con vibración frecuentemente implicados en el Raynaud ocupacional14. La actividad profesional no se había modificado en los últimos 20 años. No refirió Raynaud hasta 1998; el tiempo de aparición de los síntomas en la mayoría de los caso es entre 4 y 8 años tras la exposición al riesgo14. En la arteriografía no se objetivaron los hallazgos típicos de síndrome del martillo hipotenar como tortuosidad, trombosis, aneurismas o amputaciones de la arteria cubital14. En el inicio del proceso no se realizó cribado de conectivopatía; el paciente siguió con la misma actividad laboral, y presentaba fenómeno de Raynaud leve tras la simpatectomía. En la bibliografía no hemos encontrado casos similares, únicamente el caso de un paciente con PM con anticuerpos anti-Jo-1, que trabajó durante años expuesto al cloruro de vinilo15. Si bien no podemos ignorar que el factor mecánico traumático continuado fuese el causante de la sintomatología inicial, pensamos que el fenómeno de Raynaud pudo ser la primera manifestación de la PM.