


Sr. Director: El síndrome de TINU es una entidad rara; se han publicado únicamente unos 80 casos en la bibliografía desde su descripción en 1975 por Dobrin et al1. Es más frecuente en edades infantiles y adolescentes (edad media de 13 años) y en niñas más que en niños (proporción de 9:1 en algún estudio). Se trata de una enfermedad que se define por una tríada: a) síndrome inflamatorio sistémico, con VSG elevada, aumento de inmunoglobulinas séricas, beta-2-microglobulina, anemia y anergia cutánea; b) nefropatía con proteinuria de tipo tubular proximal, glucosuria, leucocituria, y c) uveítis anterior bilateral que suele ser no granulomatosa. Los primeros signos y síntomas (astenia, pérdida de peso, vómitos, dolor abdominal, erupciones cutáneas, atralgias, fiebre, etc.) acostumbran preceder en un mes a la tríada propia de la enfermedad. A continuación presentamos un caso de TINU con evolución y respuesta terapéutica favorables.
Varón de 14 años, sin antecedetes patológicos ni familiares de interés, que acudió al servicio de urgencias refiriendo fiebre, odinofagia, tos no productiva, cefalea, astenia, anorexia, dolor abdominal y diarrea. De acuerdo con esta sintomatología fue etiquetado de probable virasis y se recomendó tratamiento sintomático. Acudió nuevamente al cabo de 12 días por persistencia de la clínica anterior, a la que se añadían fotofobia, dolor ocular y disminución de la agudeza visual bilateral en los últimos días. Refería únicamente ingestión de paracetamol. La exploración física fue normal, excepto en la ocular, que mostró una hiperemia conjuntival y pupilas midriáticas. El diagnóstico oftalmológico fue de uveítis anterior bilateral no granulomatosa grave sin hipopión. En la analítica sanguínea destacaban VSG de 108 mm/h y PCR de 5,8 mg/dl; el hemograma objetivaba anemia normocítica normocrómica (hemoglobina, 10,6; VCM, 83,9; hematócrito, 32), 7.900 leucocitos (fórmula: 59 S/1 B/3 E/24 L/13 M), bioquímica con creatinina de 1,1 mg/dl y resto normal, incluyendo proteinograma. El estudio de orina evidenciaba una discreta proteinuria (8,8 mg/m2/h), glucosuria (+++), filtrado glomerular de 48 ml/min y el sedimento de orina con 2 hematíes/c, 3 leucos/c, sin eosinófilos. La inmunología con factor reumatoide, anticuerpos antinucleares, anti-Ro, anti-La, anticuerpos anticitoplasma de neutrófilo, enzima conversiva de la angiotensina y HLA B 27/5 fue negativa. Se llevaron a cabo serologías para herpes simple I IgG (+), herpes simple II (), CMV IgG (+), EBV IgG (+), Borrelia IgG (), Brucella (), S. Paratyphi y S. typhi () y toxoplasma (). La radiología simple y la tomografía axial computarizada (TAC) torácica fueron normales.
Durante el ingreso hospitalario presentó febrícula diaria y proteinuria discreta mantenida (entre 8 y 14 mg/m2/h), con diuresis de unos 1.500 ml/día, sin registrarse en ningún momento hipertensión arterial.
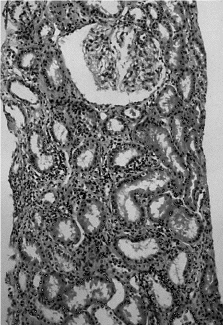
Se realizó una biopsia renal en la que el estudio óptico objetivó atrofia focal con áreas de fibrosis intersticial a nivel tubular, fibrosis y pequeños focos de infiltrado de células redondas a nivel intersticial, vasos sanguíneos y glomérulos normales. Finalmente el estudio inmunohistoquímico fue negativo para IgG, IgA, IgM y complemento, todo ello compatible con el diagnóstico anatomopatológico de una nefritis tubulointersticial (fig. 1).
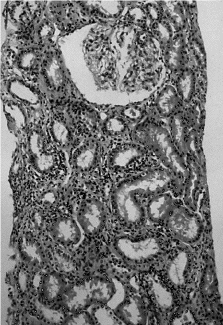
Figura 1. Biopsia renal. Atrofia focal con áreas de fibrosis intersticial a nivel tubular, fibrosis y pequeños focos de infiltrado de células redondas a nivel intersticial, vasos sanguíneos y glomérulos normales.
Se inició tratamiento sistémico intravenoso durante 4 días y tópico con corticoides. Inicialmente la respuesta clínica ocular fue baja y presentó como complicación un glaucoma. Se redujo la corticoterapia y se instauró tratamiento con ciclosporina A (CsA) vía oral a dosis inicial de 5 mg/kg/día, con lo que se consiguió un control satisfactorio de la uveítis, la normalización de los datos analíticos de nefritis y de los parámetros inflamatorios analíticos. Se continuó el tratamiento con CsA durante 18 meses, en los que mejoró la uveítis. Al cabo de dos años del diagnóstico el paciente permanece asintomático a nivel ocular, aunque con discretas lesiones residuales a la exploración oftalmológica, y no habiendo presentado tampoco más recurrencias del trastorno renal (balance renal normal, con filtrado glomerular de 114,5 ml/min).
La etiología del síndrome de TINU es desconocida, en su etiopatogenia autoinmune desempeña un papel fundamental la disfunción inmune de las células T. Se desconoce el punto de partida de la alteración inicial, pero se han implicado agentes infecciosos como "triggers": EBV, Streptococcus, Mycoplasma, Chlamydia, toxoplasma, etc.2, además de diversos fármacos, como antiinflamatorios no esteroides (AINE), antibióticos, diuréticos, entre otros. Se produce una reacción autoinmune, con presencia de receptores para IL-2 en los linfocitos del infiltrado que justificaría las lesiones tubulointersticiales renales y oculares3. Inmunológicamente se halla una situación parecida a la de la sarcoidosis, con depresión funcional específica de linfocitos TH2, que conduce a una situación paradójica de hiperreactividad celular con producción de granulomas y una hiporreactividad celular periférica con hipergammaglobulinemia4. Se ha detectado la presencia de anticuerpos anticitoplasma de neutrófilo (ANCA) en probable relación con la presencia de granulomas5.
En el estudio con microscopia óptica del tejido renal se hallan glomérulos normales o con mínimas lesiones (aumento e hipercelularidad del mesangio) y a nivel tubular predomina un infiltrado intersticial linfoplasmocitoario con o sin eosinófilos que afecta a los túbulos proximales, y en algún caso se halla una fibrosis intersticial6.
El estudio inmunohistoquímico objetiva un infiltrado intersticial de linfocitos, sobre todo de tipo T (especialmente CD4+ y en algunos estudios CD8+), macrófagos de memoria activados, neutrófilos y algún eosinófilo4. No se ha demostrado una alteración inmunológica clara (ausencia de anticuerpos en la membrana basal y estudios negativos de inmunofluorescencia) en los tejidos7.
Clínicamente lo más frecuente es una tubulopatía funcional parecida al síndrome de Fanconi o también una tubulopatía lesional con proteinuria, cilindruria, microhematuria y aumento de ß2-microglobulina, dando lugar a una insuficiencia renal moderada de evolución generalmente favorable6.
La afectación ocular más frecuente es una uveítis anterior, en algún caso posterior, o incluso puede hallarse una panuveítis, generalmente no granulomatosa, aunque se han descrito casos de granulomatosa. La evolución habitual tiende a la recidiva y es favorable la respuesta al tratamiento tópico7,8. Se ha descrito alguna complicación local como una coroiditis multifocal bilateral9.
En la mayoría de los casos no existe correlación evolutiva entre la clínica renal y la ocular. La uveítis acostumbra recidivar después de la curación de nefropatía y no está asociada al deterioro de la función renal. No se ha descrito la asociación con otra patología sistémica, aunque se conoce la existencia de nefritis tubulointersticial en otras enfermedades autoinmunes (síndrome de Sjögren, enfermedad de Behçet, lupus eritematoso sistémico, sarcoidosis, etc.)10. Sí se ha descrito la presencia de granulomas en la médula ósea y los ganglios linfáticos1,4 o en el tejido hepático5.
Inicialmente se indica el tratamiento con corticoides: prednisona (0,5-1 mg/kg/día) vía oral durante 6 a 9 meses, con respuestas importantes de la función renal y de la uveítis3. En ocasiones, el tratamiento de la uveítis debe completarse con la administración de corticoides intravítreos. En los casos en los que no se obtiene respuesta se recomienda el uso de inmunosupresores (metotrexato, azatioprina o CsA)11. El tratamiento con CsA, a las dosis recomendadas de 3,5 a 5 mg/kg/día, ha sido previamente descrito como satisfactorio4. Se ha publicado algún caso de tratamiento con CsA más corticoides vía oral con práctica resolución a los 6 meses6. Las terapias basadas en citocinas, receptores de citocinas e inmunoglobulinas intravenosas constituyen nuevas líneas de tratamiento de este síndrome12.
Nuestro caso es de un síndrome de TINU tratado con CsA más corticoides tópicos y sistémicos con una respuesta favorable, similar a la respuesta al tratamiento de la mayoría de casos revisados en la bibliografía.
Hemos creído interesante describir este caso aquí, porque consideramos que tiene importancia el diagnóstico en fases iniciales por la completa reversibilidad de la nefropatía, aunque de hecho se trata de una patología sistémica infrecuente y que en la mayoría de casos es vista por los especialistas en pediatría.