


Las lesiones ocupantes de espacio que podemos encontrar en la pared abdominal, aunque poco frecuentes, pueden tener etiologías muy diferentes. En esta revisión abordaremos el diagnóstico clínico y las opciones terapéuticas de las lesiones ocupantes de espacio más frecuentes en la pared abdominal desde el punto de vista del oncólogo médico, y por tanto, nos limitaremos a los tumores que pueden asentar en la pared abdominal, tanto primarios como metastásicos.
Space-occupying lesions of the abdominal wall arise from a large number of aetiologies. This review will describe the clinical diagnosis and the treatment approach of the most common causes of abdominal wall masses, from the Medical Oncologist view.
Los tumores de la pared abdominal suelen resultar un desafío diagnóstico y terapéutico, debido a la escasez de casos y a los pocos datos existentes en la bibliografía científica. La rareza de un tumor puede definirse en función de su prevalencia, su localización o su tipo histológico. Si tenemos en cuenta, además, que la caracterización molecular de los tumores es cada vez más compleja, y que dentro de lo que se consideraba antes un solo tipo tumoral ahora pueden establecerse subgrupos con entidad clínica, terapéutica y pronóstica propia, el resultado final es que cada vez hay más posibilidades de considerar raro un tumor, basándonos en su prevalencia.
El Project Surveillance of Rare Cancers in Europe considera como raros aquellos tumores con una incidencia inferior a 6 casos/100000 habitantes/año1.
Los tumores que encontramos en la pared abdominal los podemos dividir en dos grandes grupos: tumores primarios (51.3%) y tumores secundarios por invasión de tumores primarios intraabdominales o por implantes parietales metastásicos (48.7%)2.
Tumores primariosDentro de los tumores primarios, y siguiendo el orden de frecuencia, nos podemos encontrar los siguientes tumores:
Tumor desmoide o fibromatosis agresivaEs un tumor benigno de los tejidos blandos, que se origina en las estructuras musculoaponeuróticas de la pared abdominal. Se trata de un tumor de gran agresividad local, con gran tendencia a recidivar localmente, pero con poca capacidad de metastatizar.
Se diagnostican alrededor de 3 casos/1000000 habitantes/año, correspondiendo a menos del 3% de todos los tumores de partes blandas y al 0.03% de todas las neoplasias. Representa, aproximadamente, un 45% del total de las neoplasias de pared abdominal y un 50% de todos los tumores desmoides3–6.
El mecanismo etiopatogénico exacto se desconoce, pero parece tener un origen multifactorial en el que estarían implicados factores genéticos y hormonales. En los casos esporádicos tiende a darse en mujeres jóvenes, durante el embarazo o posparto, lo que sugiere factores hormonales en su desarrollo. También se ha visto asociado al síndrome de Gardner o poliposis adenomatosa familiar (FAP) en un 6-10%7.
En cerca del 87% de los casos esporádicos se han encontrado mutaciones somáticas en el gen CTNNB1 (3q21), que codifica la β-catenina8. En casos con poliposis adenomatosa familiar (PAF), los tumores desmoides se han asociado a mutaciones en el gen supresor de tumores APC (5q21-q22), que codifica la proteína de la poliposis adenomatosa del colon.
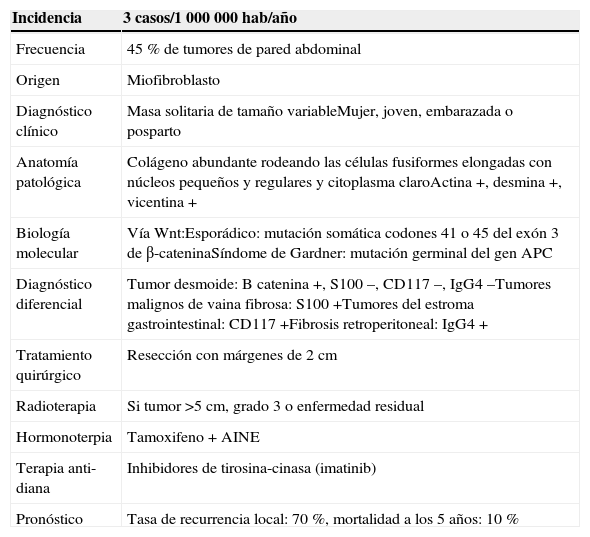
El diagnóstico inicial se basa en la clínica (presencia de una masa solitaria, dura y bien delimitada) y en técnicas de imagen con tomografía axial computarizada y/o resonancia magnética, que ponen de manifiesto la presencia de una masa infiltrante. La confirmación diagnóstica se realiza mediante una biopsia del tumor, que muestra colágeno abundante rodeando las células fusiformes elongadas con núcleos pequeños y regulares y citoplasma claro. El examen inmunohistológico muestra la expresión de los marcadores musculares (actina, desmina, vimentina) y la ausencia de CD34 (tabla 1).
Características clínicas, anatomopatológicas, terapéuticas y pronósticas del tumor desmoide
| Incidencia | 3 casos/1000000 hab/año |
|---|---|
| Frecuencia | 45% de tumores de pared abdominal |
| Origen | Miofibroblasto |
| Diagnóstico clínico | Masa solitaria de tamaño variableMujer, joven, embarazada o posparto |
| Anatomía patológica | Colágeno abundante rodeando las células fusiformes elongadas con núcleos pequeños y regulares y citoplasma claroActina+, desmina+, vicentina+ |
| Biología molecular | Vía Wnt:Esporádico: mutación somática codones 41 o 45 del exón 3 de β-cateninaSíndome de Gardner: mutación germinal del gen APC |
| Diagnóstico diferencial | Tumor desmoide: B catenina+, S100–, CD117–, IgG4–Tumores malignos de vaina fibrosa: S100+Tumores del estroma gastrointestinal: CD117+Fibrosis retroperitoneal: IgG4+ |
| Tratamiento quirúrgico | Resección con márgenes de 2cm |
| Radioterapia | Si tumor>5cm, grado 3 o enfermedad residual |
| Hormonoterpia | Tamoxifeno+AINE |
| Terapia anti-diana | Inhibidores de tirosina-cinasa (imatinib) |
| Pronóstico | Tasa de recurrencia local: 70%, mortalidad a los 5 años: 10% |
El tratamiento de elección, y único tratamiento curativo, es la resección quirúrgica. Debe realizarse la extirpación completa del tumor con márgenes libres de 2cm. No obstante, el riesgo de recaída de pacientes con los márgenes positivos es controvertido9,10. La radioterapia se administra tras la cirugía para disminuir el riesgo de recurrencia en pacientes que tienen márgenes quirúrgicos positivos, o en pacientes irresecables, aunque en casos seleccionados, con radioterapia se puede obtener un buen control local en el 75% de los casos11.
El tratamiento sistémico estaría indicado solo en casos de enfermedad avanzada o irresecables o en aquellos casos donde se intente obtener una respuesta rápida.
Existen varias opciones de tratamiento para tumores desmoides:
a) Imatinib: es un inhibidor de la tirosina cinasa, y uno de los fármacos más utilizados en el tratamiento de estos tumores, a pesar de que los resultados no son muy alentadores, ya que es el fármaco del que se dispone de más datos y que presenta mayor tasa de respuestas, alguna de ellas duraderas.
En un estudio fase ii multicéntrico, realizado por el grupo Sarcoma Alliance for Research through Collaboration, que incluyó 51 pacientes, el imatinib presentó una tasa de respuestas del 6% y una tasa de supervivencia libre de progresión al año del 66% en pacientes con tumores desmoides irresecables12.
En otro estudio fase ii del Grupo Francés de Sarcomas, que incluyó 40 pacientes tratados con imatinib (con un seguimiento de 32 meses), se observó una tasa de supervivencia libre de progresión y de supervivencia global, a los 2 años, del 55% y 95%, respectivamente. El índice de respuestas objetivas (RO) fue de 12%: concretamente, 3% respuestas completas y 9% respuestas parciales (RP)9.
La dosis de imatinib es, al igual que en los tumores GIST, de 400mg/día, administrados hasta la progresión. En el momento de la progresión se puede aumentar a dosis de 600-800mg/día. El fármaco fue bien tolerado y de pocos efectos secundarios relevantes. Los abandonos de tratamiento por toxicidad fueron también muy pocos.
b) Los antiinflamatorios no esteroideos (sulindac, celecoxib), con o sin tratamiento hormonal (tamoxifeno, toremifeno), representan otra opción terapéutica para los pacientes resistentes o que no toleran imatinib. Se ha obtenido estabilización de la enfermedad en la mitad de los casos, aunque generalmente de corta duración (inferior al año).
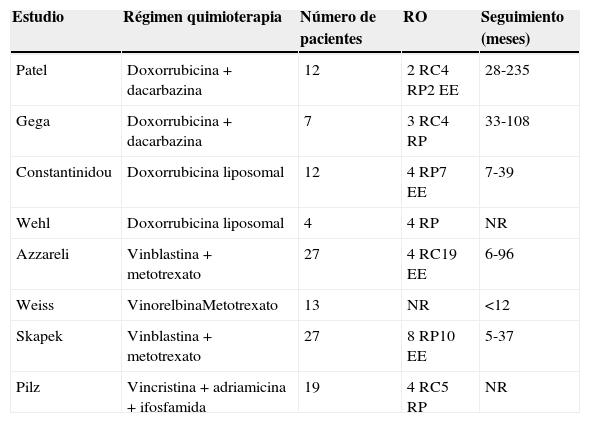
c) Algunos pacientes en progresión sintomática e inoperable se pueden beneficiar del tratamiento quimioterápico paliativo. Se han publicado estudios en los que se han observado respuestas importantes al tratamiento con fármacos citostáticos (adriamicina, DTIC). No obstante, esos estudios son poco relevantes, dado el escaso número de pacientes y la corta duración de la respuesta (tabla 2).
Regímenes de quimioterapia para pacientes con tumor desmoide avanzado
| Estudio | Régimen quimioterapia | Número de pacientes | RO | Seguimiento (meses) |
|---|---|---|---|---|
| Patel | Doxorrubicina+dacarbazina | 12 | 2 RC4 RP2 EE | 28-235 |
| Gega | Doxorrubicina+dacarbazina | 7 | 3 RC4 RP | 33-108 |
| Constantinidou | Doxorrubicina liposomal | 12 | 4 RP7 EE | 7-39 |
| Wehl | Doxorrubicina liposomal | 4 | 4 RP | NR |
| Azzareli | Vinblastina+metotrexato | 27 | 4 RC19 EE | 6-96 |
| Weiss | VinorelbinaMetotrexato | 13 | NR | <12 |
| Skapek | Vinblastina+metotrexato | 27 | 8 RP10 EE | 5-37 |
| Pilz | Vincristina+adriamicina+ifosfamida | 19 | 4 RC5 RP | NR |
EE: enfermedad estable; NR: no disponible; RC: respuesta completa; RO: respuestas objetivas; RP: respuesta parcial; SG: supervivencia global; SLP: supervivencia libre de progresión.
Fuente: Kasper B, Strobel P, Hohenberger P. Desmoid tumors: clinical features and treatment options for advanced disease. The Oncologist. 2011;16:682-93.
En pacientes con FAP el tumor desmoide constituye la primera causa de mortalidad en pacientes a los que se ha realizado proctocolectomía. En una reciente revisión de 79 pacientes con FAP, que presentaban 149 tumores desmoides (59% únicos y 41% multiples) se analizaron los resultados respecto a los distintos tratamientos. Se realizó cirugía solo en 11 pacientes, mientras que en 31 se realizó tratamiento combinado y en 20 no se realizó ningún tratamiento, por estar la enfermedad estabilizada o en regresión espontánea.
El índice de respuestas a los diversos tratamientos fue el siguiente: quimioterapia (77%) (10/13), sulindac y tamoxifeno (50%) (6/12), tamoxifeno (40%) (6/15), imatinib (36%) (4/11) y sulindac (28%) (8/29). Entre los pacientes operados se obtuvo RO 26 (62%). Tras una mediana de seguimiento de 81 meses, 8 pacientes murieron por su enfermedad y 6 por otras causas. La supervivencia global y específica a los 20 años fue del 52% y 79%, respectivamente13.
Dada su complejidad, el tratamiento debe ser programado por un equipo multidisciplinar que valore individualmente a cada paciente y que elabore la estrategia terapéutica en cada caso.
Dado el comportamiento localmente invasivo, asociado a una alta tasa de recurrencia pero con bajo potencial metastásico, el seguimiento de los pacientes con tumor desmoide se debe realizar con controles de imagen cada 4-6 meses durante los 2-3 primeros años, y posteriormente, cada año.
Sarcomas de partes blandasLos sarcomas de partes blandas representan un grupo muy heterogéneo de tumores, constituidos por una amplia variedad de subtipos histológicos, que pueden afectar a los tejidos conectivos extraesqueléticos, incluyendo músculos, tendones, grasa, fascias y membranas sinoviales, nervios periféricos y endotelio de los vasos sanguíneos y linfáticos, así como las membranas mesoteliales. Los sarcomas de partes blandas constituyen aproximadamente el 1% de todas las neoplasias malignas en adultos, con una incidencia de 2-3 casos/100000 habitantes/año. En la pared abdominal presenta una menor incidencia que los tumores desmoides, pero hay que tenerlos en cuenta para realizar un diagnóstico diferencial.
La forma de presentación más frecuente de los sarcomas de partes blandas al nivel de la pared abdominal es el crecimiento gradual e indoloro de una masa, por lo general dura, mal circunscrita, de bordes difusos y adherida a tejidos vecinos14,15.
La valoración del paciente debe incluir estudios radiológicos con tomografía computarizada y resonancia magnética, y estudio histológico para tipificar el subtipo histológico y la correcta gradación histológica según el Grupo Francés de Sarcomas16.
El tratamiento de los sarcomas de partes blandas depende de una serie de factores, como el tipo histológico, el grado y el estadio. En general, en estadios localizados, la mejor opción terapéutica es la resección completa del tumor con márgenes libres (cirugía compartimental). El papel de la irradiación en el tratamiento de los sarcomas se limita a aquellos tumores de bajo grado mayores de 5cm y a braquiterapia adyuvante en los sarcomas de alto grado para alcanzar mayor control local. El tratamiento sistémico con quimioterapia estaría indicado en tumores irresecables y/o metastásicos, y los fármacos más utilizados son la adriamicina y la ifosfamida.
Los sarcomas de partes blandas más frecuentemente encontrados en la pared abdominal son:
Dermatofibrosarcoma protuberansEl dermotofibrosarcoma protuberans (DFSP) se considera un tumor fibrohistiocítico de agresividad intermedia, por su alta tasa de recurrencias locales y baja tasa para desarrollar metástasis.
Su incidencia es, aproximadamente, de 0.8 a 5 casos/1000000 habitantes/año, y afecta preferentemente a adultos jóvenes entre los 20-45 años, con un predominio en varones. La presentación clínica es como una placa o un nódulo mal definido, que por su lento crecimiento se confunde con un nódulo benigno, aunque en algunos casos adquiere una coloración marronáceo-violácea, y puede ulcerar la piel17.
A nivel molecular se relaciona en más del 90% de los casos con cromosomas supernumerarios, que consisten en secuencias amplificadas de los cromosomas 17 y 22 o en traslocaciones recíprocas entre los cromosomas 17 y 22, t (17;22). Como resultado se desarrolla la fusión del gen del factor de las plaquetas (PDGFB) con el factor del gen colágeno tipo 1A1 (COL1A1)18.
El tratamiento de elección es la resección amplia, con márgenes de 1-3cm, que incluya la resección en bloque de la piel, el tejido celular subcutáneo y la fascia muscular. Algunos autores proponen cirugía de Mohs19.
En cuanto al tratamiento sistémico, el DFSP es insensible a la quimioterapia convencional para los sarcomas de partes blandas, por lo que no debe utilizarse.
El imatinib es un inhibidor de tirosina cinasa, que tiene actividad contra el PDGFR y ha demostrado actividad contra el DFSP en pacientes localmente avanzados o metastásicos. La mayoría de los casos reportados en la bibliografía son casos aislados en series muy pequeñas. La serie más larga fue publicada por McArthur en 200520, con 10 pacientes tratados con 400mg, 2 veces al día. De 8 pacientes con enfermedad localmente avanzada, 2 obtuvieron remisión completa patológica, 2 respuestas completas radiológicas y 4 RP, y pudieron resecarse completamente. De los 2 pacientes con enfermedad metastásica, se obtuvo una RP.
Todos los pacientes que respondieron tenían traslocación t (17;22).
Los pacientes resistentes a imatinib pueden responder a esquemas de quimioterapia como los empleados para los tumores desmoides, basados en metotrexato a dosis bajas y vinblastina, aunque estas respuestas son escasas y de corta duración.
Sarcoma sinovialEl sarcoma sinovial es un tumor mesenquimal de células fusiformes. Representa el 5-10% de todos los sarcomas de partes blandas. Afecta predominantemente a jóvenes menores de 50 años, con predominio en varones. A pesar de su nombre, no se origina en el tejido sinovial de las articulaciones (el tejido de origen es desconocido). El comienzo clínico suele ser en forma de masas indoloras que presentan un crecimiento habitualmente lento, lo que puede complicar el diagnóstico definitivo21.
A nivel molecular se asocia en más del 90% de los casos a una traslocación cromosómica característica t(X;18) (p11;q11), con lo que se produce un gen de fusión SYT-STT22.
En el sarcoma sinovial la presencia de necrosis, las mitosis frecuentes y la celularidad aumentada definen una forma pobremente diferenciada con peor pronóstico y mayor frecuencia de metástasis a distancia23.
El sarcoma sinovial es un tumor quimiosensible, tanto a las antraciclinas como, sobre todo, a la ifosfamida24. En un metaanálisis de estudios (fase iii) el uso de quimioterapia en combinación demostró un mayor aumento en la supervivencia global frente a un manejo secuencial con quimioterapia en monoterapia, pero con una mayor toxicidad. La poliquimioterapia puede ser útil en aquellos pacientes en los que se desee una mayor tasa de respuestas25,26.
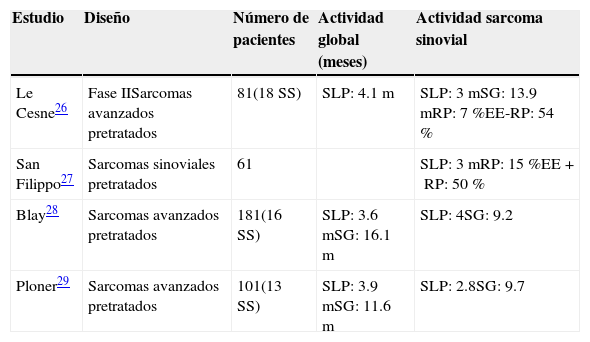
Las opciones de tratamiento tras la progresión a las antraciclinas e ifosfamida son escasas. La trabectidina (un alcaloide que se une al surco menor del ADN y bloquea la proliferación celular, con lo que detiene el ciclo celular) ha mostrado eficacia en pacientes refractarios a antraciclinas, aunque con una tasa de RO menor del 10%. La trabectidina ha mostrado en distintos estudios retrospectivos un moderado control de la enfermedad, de forma similar a los leiomiosarcomas y liposarcomas27–30 (tabla 3).
Datos de eficacia del tratamiento con trabectidina en sarcoma sinovial (estudios retrospectivos)
| Estudio | Diseño | Número de pacientes | Actividad global (meses) | Actividad sarcoma sinovial |
|---|---|---|---|---|
| Le Cesne26 | Fase IISarcomas avanzados pretratados | 81(18 SS) | SLP: 4.1m | SLP: 3mSG: 13.9mRP: 7%EE-RP: 54% |
| San Filippo27 | Sarcomas sinoviales pretratados | 61 | SLP: 3mRP: 15%EE+RP: 50% | |
| Blay28 | Sarcomas avanzados pretratados | 181(16 SS) | SLP: 3.6mSG: 16.1m | SLP: 4SG: 9.2 |
| Ploner29 | Sarcomas avanzados pretratados | 101(13 SS) | SLP: 3.9mSG: 11.6m | SLP: 2.8SG: 9.7 |
EE: enfermedad estable; RP: respuesta parcial; SG: supervivencia global; SLP: supervivencia libre de progresión; SS: sarcoma sinovial.
El angiosarcoma tiene su origen en el endotelio de los vasos sanguíneos o linfáticos. Es un tipo de sarcoma raro que representa menos del 2% del total de los sarcomas de partes blandas. Se calcula que su incidencia es de 0.1/100000 habitantes/año.
Puede aparecer a cualquier edad, y en el 30% de los casos se asocia a otras enfermedades, como a neurofibromatosis. Aunque la etiopatogenia es desconocida, el angiosarcoma se ha relacionado con la administración de radioterapia previa y otras situaciones médicas, como la presencia de «materiales extraños».
En general, se caracteriza por presentar un comportamiento agresivo, que varía en función de la localización primaria y del grado histológico. Son frecuentes las metástasis pulmonares, ganglionares, óseas y en partes blandas. Presenta una supervivencia global a los 5 años del 31%31,32.
La mejor opción terapéutica es conseguir la resección completa del tumor, con márgenes libres y resección de las metástasis, si es posible33.
Cuando existe enfermedad avanzada o metastásica, e irresecable, el tratamiento con quimioterápicos ofrece un beneficio clínico evidente en el 60-70% de los casos. Aunque no existen estudios aleatorizados, la poliquimioterapia no parece aportar beneficios en índices de respuestas ni en supervivencia frente al uso de monoterapia, según se deduce de series aisladas. Los fármacos más utilizados son:
- -
Adriamicina, que obtiene un índice de respuestas de aproximadamente un 30%, con una mediana en la supervivencia libre de progresión de aproximadamente 6 meses34.
- -
Paclitaxel, que obtiene unas tasas de respuesta del 55% (con estabilización en el 33% de los pacientes) y progresión en el 17%, con una supervivencia libre de progresión de 5 meses. Además, independientemente de que se haya utilizado en primera línea o en sucesivas, el porcentaje de respuestas se mantiene invariable35.
- -
Gemcitabina, con tasas de respuestas próximas al 75%, con beneficio clínico del 76%. Combinaciones de gemcitabina con DTIC o docetaxel también han mostrado su eficacia36,37.
- -
Antiangiogénicos como sunitinib y sorafenib, que han mostrado unos índices de respuesta de aproximadamente un 15%, aunque se ha reportado resultados de una serie de 3 pacientes tratados con sorafenib en los que se observan respuestas prolongadas38.
Los liposarcomas representan casi el 20% de los sarcomas de partes blandas, con un pico de incidencia entre los 40 y los 60 años. Se trata de tumores con una alta tendencia a la recidiva local (40-50% a los 3 años), debido, sobre todo, a la presencia de una pseudocápsula tumoral que en muchas ocasiones se encuentra vinculada a estructuras vecinas irresecables39,40.
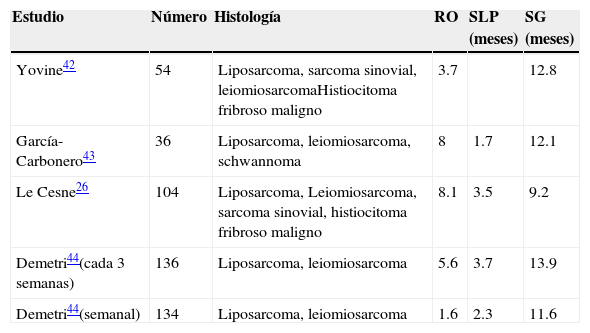
La cirugía seguida de quimioterapia adyuvante con antraciclinas constituye el tratamiento de elección. Sin embargo, la frecuencia de las recidivas hace que se planteen alternativas de tratamiento de rescate. La trabectidina, como tratamiento de segunda línea o de rescate, se ha evaluado en el sarcoma de partes blandas en varios ensayos (fase ii), con respuestas, sobre todo, en liposarcomas y leiomiosarcomas41,42.
En el estudio de Le Cesne participaron 104 pacientes. La tasa de RO fue del 8.1%, y el beneficio clínico en el leiomiosarcoma, del 56%. Se alcanzó una mediana de supervivencia libre de progresión de 3.5 meses, y el 29% de los pacientes se mantuvo sin progresión a los 6 meses. La mediana de supervivencia global fue de 9.2 meses26.
En el estudio de Yovine (fase ii) participaron 54 pacientes, con una mediana de 3 ciclos de tratamiento, y se obtuvo una tasa de RO del 3.7%. Se alcanzó una mediana de supervivencia libre de progresión de 1.9 meses, y el 24% de los pacientes se mantuvo sin progresión a los 6 meses. La mediana de supervivencia global fue de 12.8 meses42.
En el estudio de García-Carbonero se incluyó a 36 pacientes, con una tasa de respuesta global del 8% (1-13%). La mediana de tiempo hasta la progresión fue de 1.7 meses, y la mediana de supervivencia global, de 12.1 meses43.
El único estudio aleatorizado y multicéntrico, con revisión independiente, se realizó en 270 pacientes con liposarcoma y leiomiosarcoma avanzado, en progresión a antraciclinas e ifosfamida, que fueron aleatorizados en 2 grupos para recibir 1.5mg/m2 en infusión continua durante 24h cada 3 semanas o 0.58mg/m2 en 3 horas, los días 1, 8 y 15, cada 4 semanas. La mediana de supervivencia libre de progresión fue de 3.3 y 2.3 meses, respectivamente. La mediana de supervivencia global fue de 13.9 frente a 11.8 meses. La tasa de respuesta global, según criterios RECIST, fue del 5.6% y del 1.6%, respectivamente. Este estudio muestra que el régimen de 24h cada 3 semanas fue superior44 (tabla 4).
Resumen fase ii en pacientes con sarcoma de partes blandas refractarios a antraciclinas tratados con trabectidina
| Estudio | Número | Histología | RO | SLP (meses) | SG (meses) |
|---|---|---|---|---|---|
| Yovine42 | 54 | Liposarcoma, sarcoma sinovial, leiomiosarcomaHistiocitoma fribroso maligno | 3.7 | 12.8 | |
| García-Carbonero43 | 36 | Liposarcoma, leiomiosarcoma, schwannoma | 8 | 1.7 | 12.1 |
| Le Cesne26 | 104 | Liposarcoma, Leiomiosarcoma, sarcoma sinovial, histiocitoma fribroso maligno | 8.1 | 3.5 | 9.2 |
| Demetri44(cada 3 semanas) | 136 | Liposarcoma, leiomiosarcoma | 5.6 | 3.7 | 13.9 |
| Demetri44(semanal) | 134 | Liposarcoma, leiomiosarcoma | 1.6 | 2.3 | 11.6 |
RO: respuestas objetivas; SG: supervivencia global; SLP: supervivencia libre de progresión.
Un metaanálisis con 14 estudios clínicos aleatorizados (fase ii), que comparaba, tras el tratamiento local, la quimioterapia adyuvante con esquemas que contenían doxorrubicina frente a observación, mostraba una ventaja significativa del 10% en la supervivencia libre de recidiva a favor del tratamiento adyuvante, y del 4% en la supervivencia global, aunque esta diferencia no fue estadísticamente significativa45.
Posteriormente se publicó una actualización de este metaanálisis con 18 ensayos aleatorizados que incluían 1953 pacientes, y se observó un efecto significativo, aunque marginal, sobre las recaídas locales (p=0.02) y sobre la supervivencia global en pacientes tratados con la combinación de adriamicina e ifosfamida (p=0.01)46.
A pesar de estos datos, en la actualidad, dada la toxicidad de la quimioterapia, no se recomienda la administración quimioterapia adyuvante de forma estándar (nivel de recomendación IIC). Esta debe desaconsejarse especialmente en los sarcomas de grado bajo, en los de tamaño menor de 5cm.
Quimioterapia neoadyuvante y quimiorradioterapia neoadyuvanteVarios estudios han analizado el impacto de la quimioterapia neoadyuvante en sarcomas de alto riesgo de recaída, habiéndose observado que la eficacia era superior a la observada en la enfermedad diseminada. Sin embargo, en un ensayo aleatorizado la quimioterapia neoadyuvante no demostró beneficio en comparación con la cirugía sola47.
Otros dos estudios han explorado el tratamiento neoadyuvante con quimioterapia, intercalada con la radioterapia, para evitar la toxicidad de la adriamicina. En el primero de ellos, un estudio fase i, de 31 pacientes tratados, se pudo realizar resección completa en 26 casos, y la mediana de necrosis tumoral fue del 95%; sin embargo, tanto la toxicidad hematológica como local fue importante en todos los casos48. El otro es un estudio fase ii, que incluyó 66 pacientes. Se trataron con quimioterapia (adriamicina, ifosfamida y DTIC), radioterapia y cirugía. Se obtuvo RO, tras la cirugía, en 58 pacientes, y 3 tenían R1. La supervivencia a los 5 años fue superior a la esperada (71%). El 19% presentó toxicidad severa, incluyendo 3 muertes49.
Actualmente, la quimioterapia neoadyuvante solo se considera como una estrategia en investigación en pacientes con enfermedad resecable.
Quimioterapia para enfermedad avanzadaLos pacientes con enfermedad avanzada y/o metastásica y con buen estado general se pueden beneficiar del tratamiento sistémico con quimioterapia paliativa. La respuesta a los tratamientos dependerá fundamentalmente del tipo histológico. Fundamentalmente, existen dos grupos de fármacos con actividad:
- -
Antraciclinas: el fármaco más utilizado es la adriamicina, a dosis de 75mg/m2, cada 21 días, con una actividad confirmada en la mayoría de estudios que varía entre el 9% y el 23% de RO50.
- -
Alquilantes: ciclofosfamida y, sobre todo, ifosfamida. La ifosfamida administrada a dosis altas (superiores a 10g/m2) proporciona, como agente único, hasta un 40% de RO51.
Los esquemas de poliquimioterapia no han demostrado una superioridad sobre la monoquimioterapia, en términos de supervivencia, con una mayor toxicidad52.
Invasión de tumores primarios intraabdominales e implantes parietales metastáticosLos implantes tumorales metastásicos y la invasión directa de tumores primarios intraabdominales en la pared abdominal pueden deberse a tumores de localizaciones y etiología muy diversas.
La incidencia de metástasis de cualquier neoplasia en la pared abdominal es aproximadamente de 0.7% a 9% en ambos sexos53. La mayoría de los casos reportados son metástasis de neoplasias de origen colónico.
El crecimiento tumoral en el sitio de entrada de la vía laparoscópica durante la cirugía oncológica tiene una incidencia del 1% al 2% de los procedimientos en presencia de neoplasias intraabdominales (tasa comparable a las metástasis sobre la herida quirúrgica en la cirugía tumoral abierta). La incidencia de implantes en el lugar de entrada del trocar durante una cirugía oncológica por vía laparoscópica de carcinoma colorrectal es del 0.7-1.3%54.
La etiología parece ser multifactorial, desde la implantación directa de las células tumorales a la diseminación hematógena. Se han sugerido una serie de medidas preventivas, como el lavado de la cavidad peritoneal con agentes citotóxicos y/o la instilación de estos en las heridas de los trocares, a fin de evitar el crecimiento celular. Se puede considerar la escisión del tejido que rodea el orificio de la puerta de laparoscopia como alternativa de prevención.
Clínicamente se manifiesta como una masa dura, dolorosa, de crecimiento progresivo y evidente en las exploraciones radiológicas complementarias. El tratamiento es la resección en bloque de la masa y la pared abdominal en todo su espesor.
En general, el tratamiento de elección de las metástasis en la pared abdominal y de las infiltraciones por contigüidad por tumores intraabdominales es la resección completa, si es posible, del tumor, seguida de una quimioterapia sistémica específica para cada tumor. Antes de iniciar el tratamiento debe realizarse un estudio de extensión completo para descartar la presencia de metástasis en otros órganos.
ConclusiónExisten pocos datos epidemiológicos sobre la incidencia y la prevalencia de los tumores de la pared abdominal en todo el mundo, y sobre el impacto en la morbilidad y la supervivencia que pueden causar.
Desde el punto de vista clínico, los tumores de la pared abdominal constituyen un grupo heterogéneo de enfermedades relativamente poco frecuentes en la práctica clínica habitual. Por todo ello, es necesario un enfoque multidisciplinar en su abordaje tanto diagnóstico como terapéutico, de manera que se exponga el conocimiento y la experiencia de todos los especialistas implicados en su diagnóstico y tratamiento. No cabe ninguna duda de que los avances en biología tumoral y genética de estos tumores, la identificación de nuevas dianas moleculares y el desarrollo de nuevos fármacos producirán una mejoría en las perspectivas de tratamiento de estos pacientes.
Conflicto de interesesNo existe conflicto de intereses.
