




Fusarium moniliforme is a phytopathogenic facultative fungus with a cosmopolitan distribution in all types of climates, and has a wide host range, including, among others, bean, rice, wheat and sorghum crops. There is a current lack of knowledge regarding the potential of these fungi, so it is considered to be of great importance to obtain information related to the biological activity of extracts and secondary metabolites.
AimsAn evaluation of the role of methanol:chloroform extract of F. moniliforme in the production of inflammatory cytokines and their cytotoxic activity.
MethodsThe production of nitric oxide was analyzed by the Griess method, the production of cytokines using ELISA, and the effects of the extract on cell cycle and induction of apoptosis by flow cytometry.
ResultsThe extract of F. moniliforme was seen to be able to stimulate nitric oxide (NO) production in J774A.1 cells, as well as to produce cytokines such as, IL-1β, IL-6, and TNF-α. It was also observed that the extract of F. moniliforme produces activity on cell cycle modulation and apoptosis when tested in carcinogenic cell lines.
ConclusionsThe results obtained from this study open the possibility of obtaining and identifying metabolites of the extract of F. moniliforme that can be evaluated for possible use in cancer therapy.
Fusarium moniliforme es un hongo fitopatógeno facultativo con distribución cosmopolita en todos los tipos de climas y con una gran variedad de huéspedes, como el frijol, el arroz, el trigo y el sorgo. Actualmente existe una falta de conocimiento sobre el potencial de este hongo, por lo que es de gran importancia obtener información relacionada con la actividad biológica de los extractos y sus metabolitos secundarios.
ObjetivosEn este trabajo se evaluó el papel del extracto metanol:cloroformo de F. moniliforme sobre la producción de citocinas y su actividad citotóxica sobre líneas celulares.
MétodosSe analizó la producción de óxido nítrico por el método de Griess, la producción de citocinas se evalúo por el método de ELISA y los efectos del extracto sobre el ciclo celular e inducción de apoptosis se analizó por citometría de flujo.
ResultadosEl extracto de F. moniliforme fue capaz de inducir la producción de óxido nítrico (ON) en células J774A.1, así como la producción de citocinas IL-1β, IL-6 y TNF-α. También se observó que el extracto de F. moniliforme posee actividad en la modulación del ciclo celular y en la inducción de apoptosis observada en líneas células cancerígenas.
ConclusionesLos resultados de este trabajo abren la posibilidad de obtener e identificar los metabolitos del extracto de F. moniliforme para que puedan ser evaluados en una posible terapia para el cáncer.
Fungi contain a variety of secondary and unprecedented metabolites with structural features that cause antimicrobial, antifungal, anti-inflammatory and antiparasitic effects, among others, as well as being immunomodulators.4,16 The number of species of fungi on earth is estimated to be larger than 100,000, suggesting that only 15–20% are already known.8 Even among known species, the proportion of well-investigated fungi regarding their contents in secondary metabolites, antioxidant and antimicrobial properties, is very low.9 This fact, together with the knowledge of the great potential of fungi in the production of bioactive metabolites, in addition to their use in ethno-medicine, reveals them to be a vast source of bioactive compounds.8,14 The number of fungi that have demonstrated an immunogenic and antitumor effect in animals amounts to more than 30.21 Medicinal effects have been demonstrated in species of the genera Auricularia, Flammulina, Ganoderma, Grifola, Hericium, Lentinus, Pleurotus, Trametes, Schizophyllum, and Tremella, among others.32 Several metabolites with cytotoxic and immunomodulatory bioactivity have been isolated from the already mentioned genera18; such is the case of ganodermic acids, ganoderiols and polysaccharides present in some species of Ganoderma.26
There is another large group of fungal species that parasitize any living plant material which are known as phytopathogenic fungi.1 Studies reveal the antibacterial potential of phytopathogenic fungi,29 resulting active against some bacteria.
Fusarium moniliforme is a facultative phytopathogenic fungus with cosmopolitan distribution in all climates, and has a wide host range, including crops of beans, rice, wheat and sorghum. The genus Fusarium have a large number of metabolites in their chemical composition, some of which are toxins,20,22 but there may be some potential biomedical use for these. Phytopathogenic fungi possess, like other fungal species, a large number of metabolites in their chemical composition,1 and thus represent potential options for finding new biological activity. However, Mexico has no prior knowledge that enables the detection of substances produced by these phytopathogenic fungi despite greater research related to the production of secondary metabolites from phytopathogenic fungi.30 Currently, there is a lack of knowledge regarding the potential use of these fungi, so it is considered very important to obtain information related to the biological activity of extracts and secondary metabolites. The objective of this study was to determine the cytotoxic activity and induction of inflammatory mediators of the methanol:chloroform extract of F. moniliforme.
Materials and methodsCollection and identification of F. moniliformeF. moniliforme was collected from citrus fruits (orange and lemon) cultivated at Martínez de la Torre, Veracruz, México. Sampling was performed seasonally in the summer of 2010. F. moniliforme specimens were collected and conserved following conventional mycological techniques. Taxonomic identification was reached according to Gilbertson and Ryvarden and Larsen & Cobb-Poulle.12 Approximately 30g of mycelia were cultivated in potato dextrose broth (DIFCO) for 7 days at 25°C with constant stirring at 50rpm. They were then freeze dried and vacuum stored to −35°C until extraction.
Obtaining bioactive extracts from F. moniliformeMethanol:chloroform extracts of F. moniliforme were obtained as follows: 1g of freeze-dried of the sample was mixed in 15ml of 80% (v/v) methanol, and homogenized with an Ultra-Turrax T 25 basic (IKA® WERKE, Germany); thereafter the homogenate was sonicated for 15min and centrifuged at 12,000×g at 4°C for 15min. The sample was vacuum-filtered through Whatman No. 1 filter paper. This procedure was repeated twice to ensure the maximum extraction of bioactive compounds using 20ml of 1:1 (v/v) methanol:chloroform mixture. The extracts were collected and made up to a final volume of 50ml, the solvent was then removed by evaporation under reduced pressure and the obtained dry extract was re-suspended in dimethyl sulphoxide (DMSO). Finally, all necessary dilutions were made from a stock solution (1mg/ml) to obtain the desired concentrations (0–150μg/ml), which was used for cytokines production and the determination of cytotoxic activity.
Cell linesCytotoxicity studies were carried out on several cell lines, such as epithelial Vero cells (ATCC: CCL-81), osteoblast MG-63 (ATCC: CRL-1421), breast cancer cells HBL-100 (ATCC: HBT-124), lung cancer SW 1573 (ATCC: CRL-2170), colon cancer cells WiDr (ATCC: CCL-218), adenocarcinoma cells HeLa (ATCC: CCL-2), and U937 cells (histiocytic lymphoma) (ATCC: CRL-1593.2). The cells were maintained in DMEM (Dulbecco's modified Eagle's medium) (Sigma), with the exception of the U937 cell line that was grown in RPMI 1640. The medium was supplemented with 10% fetal calf serum (GIBCO), 1% antibiotic solution (penicillin 1000IU/ml+streptomycin 250mg/ml) and 2mM l-glutamine (200mM).
In vitro stimulationMurine macrophages J774A.1 (ATCC: TIB-67) were incubated with DMEM containing 10% FCS at 37°C in 5% CO2 in 24 well plates (105 cells/ml). Cells were cultured separately in the presence of 2–15μg/ml of the extract of F. moniliforme or the medium alone. Cells and culture supernatants were collected at different times (48 and 72h), nitric oxide (NO) production and cytokine concentrations were determined. Polymyxin B sulphate (PMB) (Sigma Chemical Co.) was added to inhibit the lipopolysaccharides (LPS) present in the extract derived from the purification process. DMSO was used as a negative control in all experiments.
Cytotoxicity assay by sulphorhodamine BThe cells were seeded onto 96-well microtitre plates at a concentration of 2.5×104 cells per well, and allowed to proliferate for 24h in DMEM containing 10% fetal bovine serum. Different concentrations of extract of F. moniliforme (0, 5, 10, 15, 25, 30, 50, 70, 90, 100, and 150μg/ml) were applied to the cell monolayer in triplicate; DMSO was used as the negative control. After incubation at 37°C with 5% CO2 for 48h, the cell growth was evaluated by the sulphorhodamine B assay (SRB). The SRB assay was carried out as previously described.19 Briefly, the culture medium was suctioned prior to fixation and 75μl of 10% cold (4°C) trichloroacetic acid was gently added to the wells. Microplates were left for 30min at 4°C, washed five times with deionized water and left to dry at room temperature for at least 24h. Subsequently, 75μl 0.4% (w/v) sulphorhodamine B (Sigma) in 1% acetic acid solution was added to each well and left at room temperature for 20min. The SRB was removed and the plates were washed five times with 1% acetic acid before air-drying. Bound SRB was made soluble with 70μl 10mM un-buffered Tris-base solution, and the plates were left on a plate shaker for at least 10min. Absorbance was read at 530nm by subtracting the background measurement of 620nm. The test optical density value was defined as the mean absorbance of each individual well, minus the blank value (‘blank’ is the mean optical density of the background control wells). The results were expressed as the percentage of viability. The 50% cytotoxic concentration (CC50) was defined as the concentration that reduces the optical density (OD530) of treated cells to 50% of untreated cells.
Nitric oxide measurementNitrite accumulation, an indicator of NO synthesis, was measured in the culture medium by the Griess reaction. In brief, J774A.1 were stimulated with 2–15μg/ml of the extract of F. moniliforme, LPS from Escherichia coli (0.0111:B4, 4ng/ml) (Sigma Chemical Co.), IFN-γ (100U/ml) (Genzyme Diagnostic), or LPS plus IFN-γ, respectively. Non-stimulated cells were used as control. In some cases, NG-nitro-l-arginine methyl ester (l-NAME; 3mM) (Sigma Chemical Co.) was added separately; similarly 15μg/ml of PMB (Sigma Chemical Co.) was added to inhibit the LPS present in the extract derived from the purification process (data not shown). One hundred microlitres of the cell culture medium were mixed with 100μl of Griess reagent and incubated at room temperature for 15min. Absorbance at 540nm was determined, and nitrite concentration was calculated from a sodium nitrite standard curve.
Determination of cytokine pattern by ELISAInterleukin 1-beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) were quantified by ELISA in culture supernatants of J774A.1 under different conditions of stimulation, according to the manufacturer's protocol. Briefly, 96-well flat-bottom plates were coated overnight with a capture antibody at a final concentration of 2μg/ml, and then plates were blocked with 10% PBS-FCS, washed three times, and incubated with the cell culture supernatant samples or control antigens overnight at 4°C. After washing, the plates were incubated with the respective biotinylated anti-cytokine antibodies (R&D System) at 1μg/ml for 1h in the dark. The plates were then washed and streptavidin–alkaline phosphatase at 1:2000 was added. Plates were incubated for 30 min in the dark. After washing the plates again, 100μl of ABTS (2,2,-azino-bis (3-ethylbenzthiazoline)-6-sulphonic acid) (Zymed) were added as substrate, and the reaction was allowed to proceed for 20min at room temperature (RT); the reaction was stopped with 2% sulphuric acid, and absorbance was read at 415nm by an ELISA reader (Multiskan MS, Labsystem).
Induction of cell damageU937 cells were treated with various concentrations (1–30μg/106 cells) of extract of F. moniliforme during 2, 4, 6 and 8h. After treatment, non-adherent cells were removed, and the monolayer was trypsinized. The trypsinized cells were combined with the non-adherent cells before staining. The cells were then washed twice in phosphate-buffered saline solution (PBS), containing 0.05% azide and 1% FCS, stained with trypan blue and counted in an automatic counter (TC10 Automated Cell Counter 145-0001 BIO-RAD; USA), and viable cells were re-suspended in PBS at a concentration of 1×106/ml, ready for staining.
Propidium iodide stainingCell suspensions obtained from the culture of U937 cells were centrifuged at 1200×g at room temperature for 7–8min. The supernatants were thoroughly removed by aspiration and the tubes tapped vigorously to disperse the pellets. A sterile filtered solution of 50μg/ml propidium iodide (PI) in 10−2M Tris, pH 7.0 containing 5mM MgCl2 was added slowly. The tubes were tapped vigorously during the course of the addition to help suspend the pellets. Vortexing was avoided. One milliliter of PI solution was added for each 106 cells. After staining, the cell population was analyzed by flow cytometry.
Actinomycin D (7AAD) staining and flow cytometer7AAD (Calbiochem-Novabiochem, Nottingham, UK) was dissolved in acetone and diluted in PBS to a concentration of 200μg/ml. This was kept at −20°C and protected from light until use. A total of 100μl of 7AAD solution was added to 106 U937 cells suspended in 1ml PBS and was mixed well. The cells were stained for 20min at 4°C while protected from light and then were pelleted by centrifugation. The supernatant was removed, and the cells were re-suspended in 500μl 2% paraformaldehyde (PF) solution (Sigma). Unstained fixed cells were used as negative controls. Samples were analyzed on a CYAN-ADP cell sorter (Beckman Coulter, Brea, CA.) within 30min of fixation. The whole nucleated cell population was analyzed. Data on 100,000 cells was acquired and processed using Flowing software 2.5.0 (Turku Bioimaging, Turku, Finland).
Statistical analysisThe experiments were performed in three replicates for each condition and were carried out at least twice to ensure reproducibility. CC50 calculation was done with the program Prism version 5.0 using a non-linear regression analysis. One-way analysis of variance (ANOVA) and Tukey multiple comparison tests were used for statistical analyses. Statistical significance was defined when the p value was less than 0.05 (p<0.05).
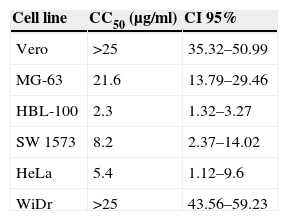
ResultsCytotoxic activity of the extract of F. moniliformeThe cytotoxic effect of the extract of F. moniliforme was evaluated on MG-63, HBL-100, SW 1573, WiDr, and HeLa cancer cell lines in vitro using the sulphorhodamine B assay. Extract concentrations ≤10μg/ml exerted a ∼50% reduction on cell viability in HBL-100, SW 1573, and HeLa cultures, whereas in HBL-100 cultures a similar cell viability loss was achieved with 2.5μg/ml of extract. Obtained CC50 values for HBL-100, SW 1573, and HeLa cell lines were 2.3μg/ml, 8.2μg/ml, and 5.4μg/ml, respectively (p<0.001) (Table 1). However, the extract was less cytotoxic toward normal human osteoblast cell line MG-63. The WiDr cells viability was not affected when treated with the extract at 15μg/ml concentration but the viability of the cells was reduced to 80% when treated at 35μg/ml (p<0.001) (Table 1). The extract was considered to possess very strong cytotoxic activity toward HBL-100 cells and moderate activity against SW 1573 and HeLa cells. A test was also performed with non-tumor cells (Vero cell line) which were unaffected by interaction with the extract.
Cytotoxic activity of the methanol:chloroform extract from Fusarium moniliforme.a
| Cell line | CC50 (μg/ml) | CI 95% |
|---|---|---|
| Vero | >25 | 35.32–50.99 |
| MG-63 | 21.6 | 13.79–29.46 |
| HBL-100 | 2.3 | 1.32–3.27 |
| SW 1573 | 8.2 | 2.37–14.02 |
| HeLa | 5.4 | 1.12–9.6 |
| WiDr | >25 | 43.56–59.23 |
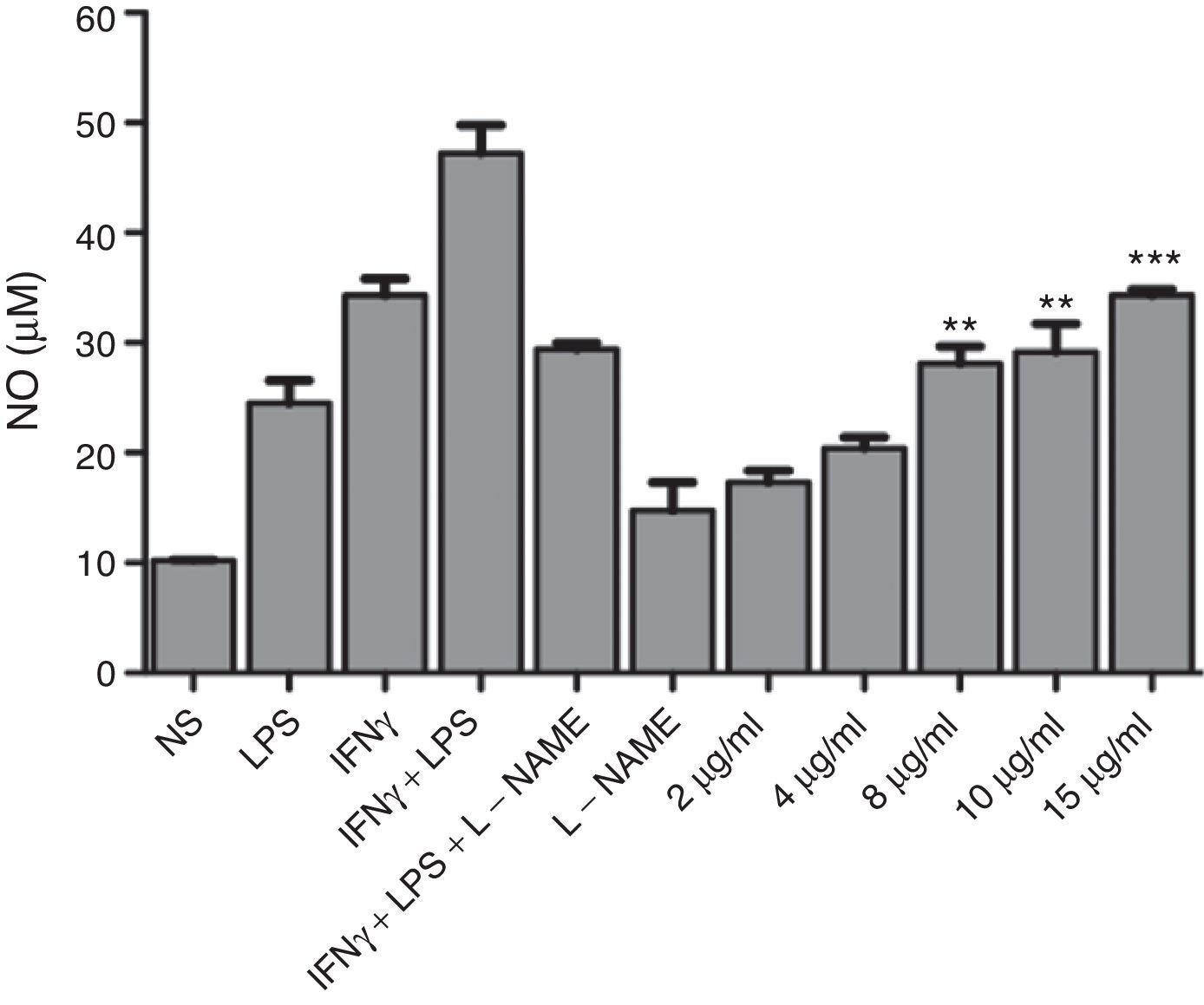
To test the ability of the extract to induce nitric oxide production, J774A.1 were stimulated with a different concentration of the extract in vitro. Result showed that extract of F. moniliforme is capable of inducing NO production in J774A.1 after 48h of stimulation (Fig. 1), and that production is inhibited by the action of the inhibitor l-NAME (data not shown). Nitrite values obtained by the stimulation with 8, 10, and 15μg/ml of the extract are statistically significant when compared with the values of non-stimulated cells (p<0.001) (Fig. 1). NO production was increased up to 72h (data not shown).

F. moniliforme extract induces NO synthesis in J774A.1. Macrophages cell line J774A.1 were stimulated at 48h with different stimuli. Cells cultured with medium alone were used as controls. Supernatants of cultured cells were harvested, and nitrite concentration was assayed. Data are expressed as means±SD and are representative of three independent experiments. *, **, *** p<0.05, 0.001, 0.0001 respectively compared with unstimulated cells and controls.
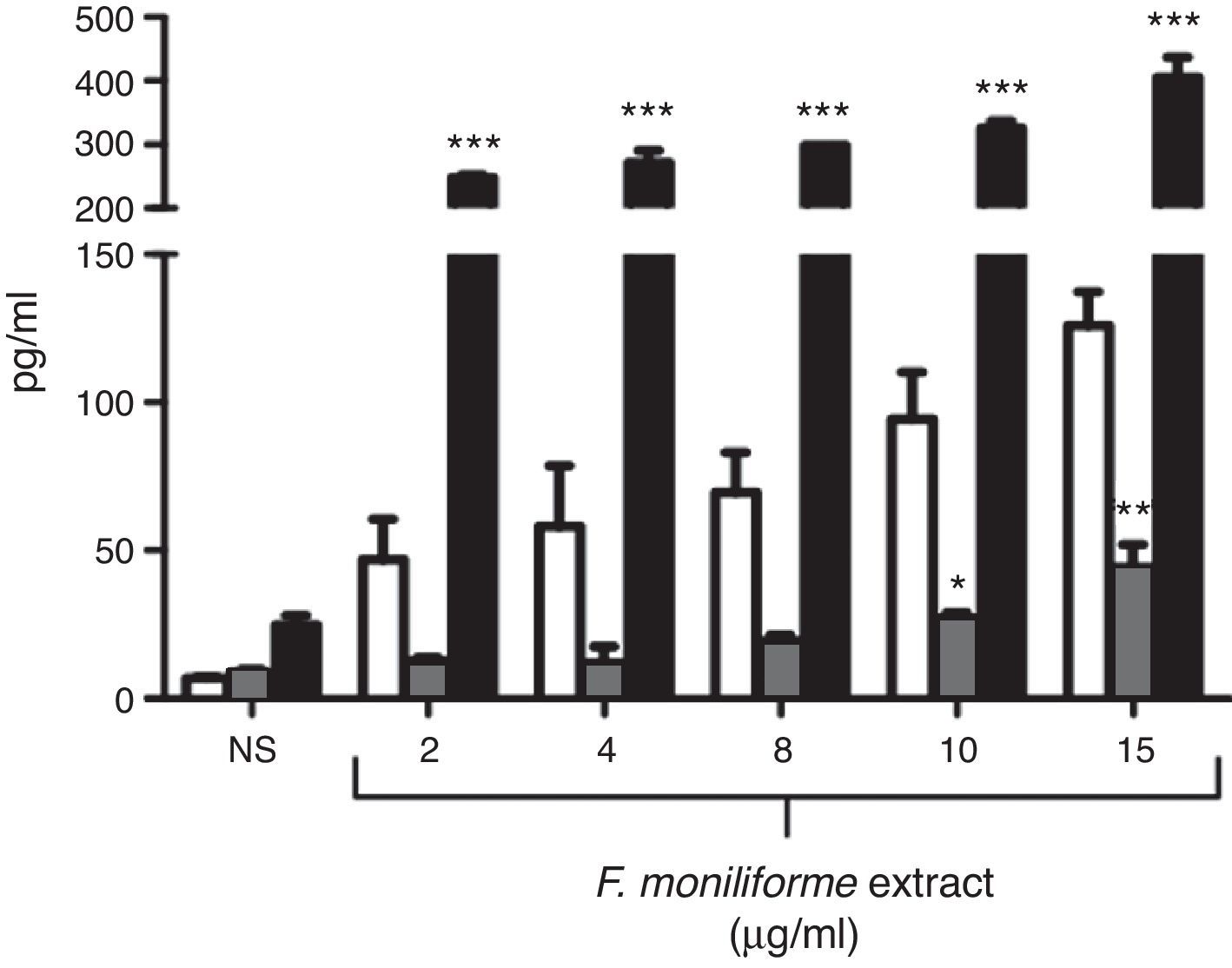
Cytokine production by J774A.1 cells was stimulated by the extract of F. moniliforme. When J774A.1 cells were stimulated with the extract, the production of IL-1β, IL-6, and TNF-α significantly increased (Fig. 2). For IL-1β, the increase was observed after 48h at a concentration of 15μg/ml, while for IL-6, the increase was noticed after 48h at a concentration of 10–15μg/ml and a sustained production of TNF-α from 48h of interaction at a concentration 2–15μg/ml.
, IL-6 (checkered bars), and TNF-α (black bars). Histograms show values in pg/ml (means±SD) of three experiments run in duplicate. *, **, *** p<0.05, 0.001, and 0.0001 respectively vs. unstimulated cells.")
Profile of cytokines induced by extract of F. moniliforme in J774A.1. Macrophages from cell line J774A.1 were stimulated with the extract for 72h, and cytokines were measured in culture supernatants from cells by ELISA. IL-1β (open bars), IL-6 (checkered bars), and TNF-α (black bars). Histograms show values in pg/ml (means±SD) of three experiments run in duplicate. *, **, *** p<0.05, 0.001, and 0.0001 respectively vs. unstimulated cells.
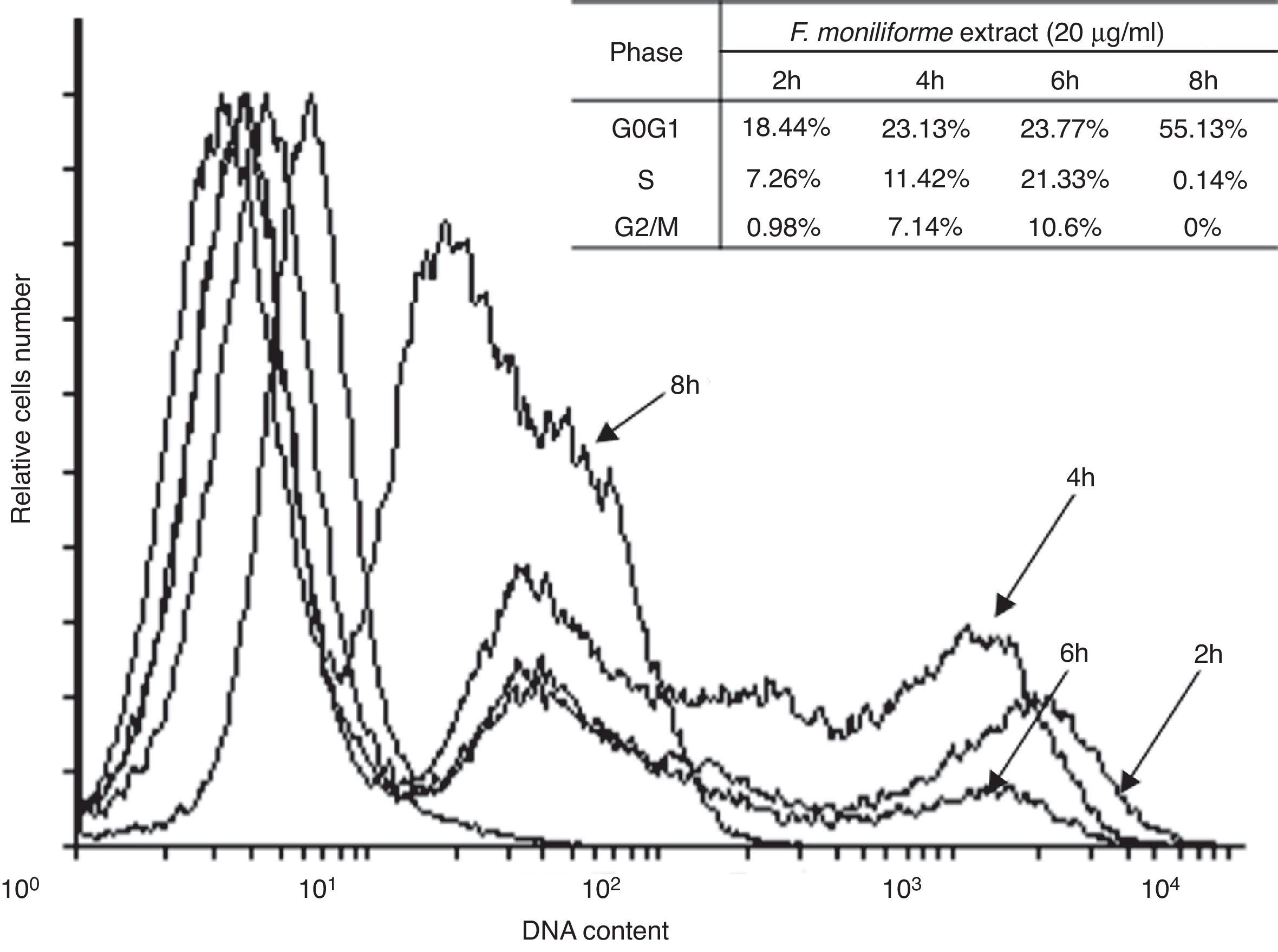
Cell cycle arrest is one of the major causes of cell growth inhibition. In order to find whether cell growth inhibition comes from cell cycle arrest at a specific phase of cell cycle or not, the cell cycle profile was analyzed using PI staining and flow cytometry analysis. Our results showed that the extract of F. moniliforme (20μg/ml) arrested the cell cycle at G0/G1 phase in a time-dependent manner (Fig. 3). After 2h of interaction with the F. moniliforme extract, the cellular population in G0/G1 phase was 18.44%, and after 4h of interaction 50% cell viability was observed. The viability of the cell population in G0/G1 phase after 8h was 55.13%. The absence of cells in the S phase and G2/M was also noted after 8h. These results strongly suggest that the extract of F. moniliforme prevents replication of DNA in these cells and that they may have a degree of cell damage (Fig. 3).

Effect of the extract of F. moniliforme on cell cycle. U937 cells were cultured with the extract at 20μg/ml for 2, 4, 6 and 8h and the cell cycle phase distribution was determined by PI staining and analyzed using flowjo software. The percentages of cell cycle phases are shown in the table.
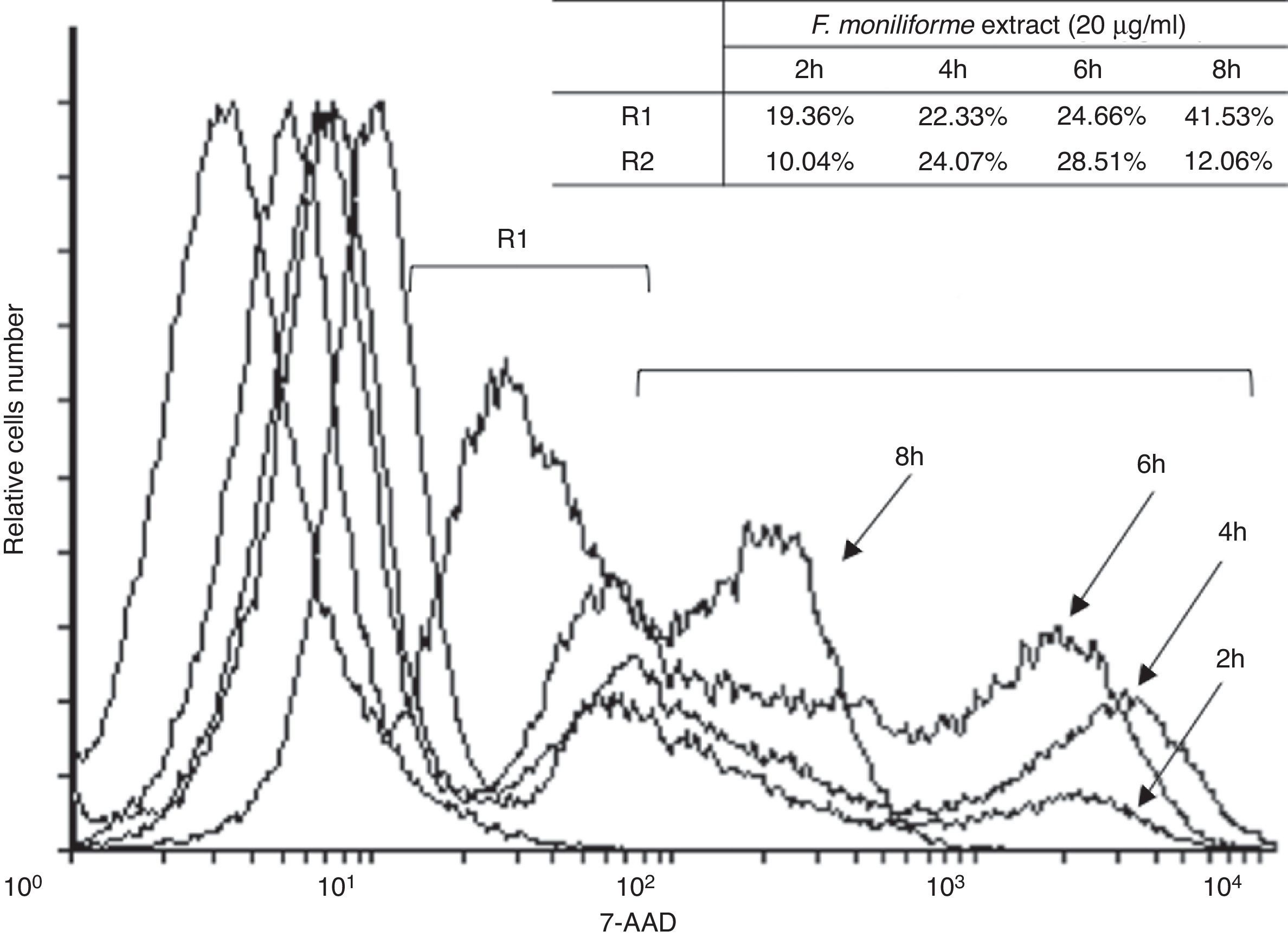
U937 cells were treated with 20μg/ml of the extract of F. moniliforme and then stained with 7AAD. The proportion of apoptotic and dead cells increased in all cases in a dose-dependent manner (Fig. 4). Based on the properties of the membrane, late apoptotic cells incorporate more 7ADD. The results show that prolonged interaction of the cells with the extract induced apoptosis of the cell population in the early stages (Fig. 4).

Flow cytometric analysis of apoptosis in U937 cells treated with extract of F. moniliforme. U937 cells were treated with 20μg/ml of extract for 2, 4, 6, and 8h, and were stained with 7-AAD. R1 corresponds to the cell population having early stages of apoptosis. R2 corresponds to the cell population that presents late stages of apoptosis or necrosis. The percentages of apoptosis are shown in the table.
Natural medicine has been used for the treatment of many diseases in the course of the entire history of mankind. Since ancient times, the beneficial effects of numerous plants in human health have been known, many of which have been used to treat and prevent various diseases.7,27 In the last decades there has been a widespread tendency to regain the use of products derived from natural sources such as plants and fungi, which are consumed as dietary supplements in a growing number of countries. These substances, which exhibit pharmacological properties in a wide spectrum of diseases have demonstrated their safety compared to chemical-synthetic drugs.6,23 The use of fungus extracts has been common practice in traditional medicine for centuries, including the treatment of cancer. In this paper, we analyzed the biological activities of an extract of a phytopathogenic fungus that has not been investigated for biomedical purposes previously. In order to correlate the cytotoxic action exhibited by the extracts of plants or other biological activities with the presence of certain chemical components, several authors report a toxicity classification according CC50 value of the extract. Extracts or compounds which showed a CC50 value of 10–25μg/ml were considered to be weak in cytotoxicity while compounds with a CC50 value of less than 5μg/ml were considered very active. Those compounds or extracts that have intermediate values between 5 and 10μg/ml of CC50 were classified as moderately active.2,17 According to the above, methanol:chloroform extract of F. moniliforme has a cytotoxic activity ranging from extremely active to moderately active (Table 1). It is worth mentioning that the comparison with another method such as MTT staining was not performed, since it has been reported that the method of sulphorhodamine B (SRB) was developed as an alternative to the MTT reduction method.25 On the other hand, the extract was able to induce the production of NO in macrophage cell line J774A.1. As it is known, NO is a free radical that, at low concentrations, acts as single signal transducer whereas at higher concentrations can act as a cytotoxic/cytostatic defensive mechanism against pathogens and possibly tumors.15 Lately, NO has emerged as a potential anti-proliferative agent capable of overcoming tumor cell resistance to conventional therapeutic agents. High concentrations of NO can induce cell death in several cell types.28 These include macrophages, thymocytes, pancreatic islets, certain neurons, and tumor cells, although the precise mechanism that determines the cellular sensitivity against NO induced apoptosis is not clearly elucidated.10 Moreover, the extract was able to induce the production of pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, which highlights the sustained production of TNF- α (Fig. 3). In addition to inducing an evident alteration in the cell cycle and cell damage induction (Fig. 4), there is evidence that cytokines IL-1β and TNF-α and their signaling pathway induce apoptosis and NO in many cells. Apoptosis, or programmed cell death, plays a critical role in the regulation of tissue homeostasis, especially in cell systems with a high turnover rate such as hematopoietic cells. Imbalances between proliferation and cell death may lead to impaired development, degenerative diseases and tumor formation.3 IL-1β signaling, which is mediated through MAPK activation of JNK and p38, can also lead to induced apoptosis in cells.24 TNF-α, along with its numerous effector functions, is a potent inducer of apoptosis. TNF-α induces apoptosis in endothelial cells via phosphorylation and down-regulation of Bcl-xL.13
In a Th1 environment, pro-inflammatory cytokines such as interleukin (IL)-1α, IL-1β, IL-6 and TNF-α enhance anti-cancer immunity. Inducing Th1-type inflammation may significantly improve immunotherapeutic strategies against cancer.19 Interestingly, cytokines did not induce apoptosis in normal cells. This may be due to the susceptibility,31 and that the levels of the receptor of cytokines are different between cancer cells and normal cells. In addition, it was found that many normal cells are not killed by TNF-α and this may be related to NF-κB trans-activation by blockade of NF-κB, which sensitizes the cell to TNF-α and augments induced apoptosis cell death.5 It has been observed that cytokines such as IL-1, TNF-α, and IL-6, originally identified as immune-regulatory molecules, could also act as regulators of pathophysiological resorption and were produced by many different cell types including osteoblasts, and IL-6 have also been shown to stimulate osteoclast differentiation and bone resorption in a synergistic manner,11 therefore, a decrease of the cytotoxic effect observed with the extract of F. moniliforme on osteoblasts may be due to the presence of IL-6 induced by the extract in MG-63 cells. Finally, there are reports showing that some polysaccharides isolated from edible or medicinal fungal species mediate an immune response by increasing the production of cytokines, NO, TNF-α, IFN-γ and antibodies which activate the effector cells.4,22,33 In recent years, we have seen a reorientation process of diagnosis and treatment of neoplasms. The current trend line for improving immunomodulatory therapies, and treatments with compounds of biological response are an effective and promising alternative in the management of cancer. Preliminary results of this research open the possibility of obtaining and identifying metabolites of the extract of F. moniliforme that can be evaluated for possible cancer therapy.
ConclusionsMethanol:chloroform extract from mycelia of F. moniliforme isolated from oranges and lemons from Martínez de la Torre, Veracruz, México, revealed considerable cytotoxic and immunogenic properties as evidenced by their activity against cancer cell lines and by the induction of cytokines and nitric oxide in murine macrophages, and shows a potential cytotoxic activity due to the inhibition of growth and cellular damage.
Conflict of interestThe authors have no conflict of interest to declare.
The authors would like to thank Ph.D. Vianney Ortíz-Navarrete for his technical assistance and Wendy del Rosío Hernández Martínez for the revision of the paper. María de la Soledad Lagunes-Castro was a recipient of a Ph.D. fellowship from CONACyT, México (249756). This work was supported by FOMIX CONACYT Gobierno del Estado de Veracruz (VER-2009-C03-128039) to ARL, and SEP-CONACYT básica-2012 (181820) to ATL.

