




Mitosporic fungi with entomopathogenic habit have been widely used as pesticides because of their potential in biological control of insect pests. Due to their predominant asexual nature, the taxonomic status of these fungi is not certain. The sexual stages (teleomorphs) of some of these genera have been discovered. The teleomorphs are ascomyceteous species. The asexual entomopathogenic fungal species were found to be closely related to each other in several molecular phylogenetic studies3,4,8,9,21,22,28,31. Further, these studies also confirmed the relation between the anamorph (asexual) and the teleomorph species. Based on these evidences, these fungi are recognized as hypocrealean ascomycetes belonging to the family Clavicipitaceae. However, no phylogenetic study of these mitosporic fungi was done including representative members of sexual Ascomycota to view their position in context to other members of Ascomycota. We took up such a study using β-tubulin and rRNA gene sequences. Many of the economically important hyphomycete entomopathogenic fungi are complex species and the boundaries between different morphologically distinguishable species are not validated. Therefore, inter- relationships between some of the important entomopathogenic genera were also investigated including a sample of isolates of complex species - Beauveria bassiana and Nomuraea rileyi, two of the five hyphomycetous asexual fungal species registered as bioinsecticides29.
Of the several genes used to serve as 'zeit gebers' or molecular clocks for studying phylogenetic relationships among organisms, the highly conserved and functionally essential β-tubulin gene has been found to be very useful for investigating relationships between fungi at all levels - from studies of complex species groups to deep (geological) time phylogenetic investigations5,8,12,16,18-20. Since it is a protein-coding gene, it has some advantages over the more often used ribosomal RNA gene sequences: the alignment of the sequences is less problematic and the third nucleotide can provide a relatively good estimate of the neutral substitution rate33. rRNA genes are also frequently used to establish ordinal relationships among fungi1,9,17. Both these gene sequences were employed in the present study.
Material and methods
Fungal sample
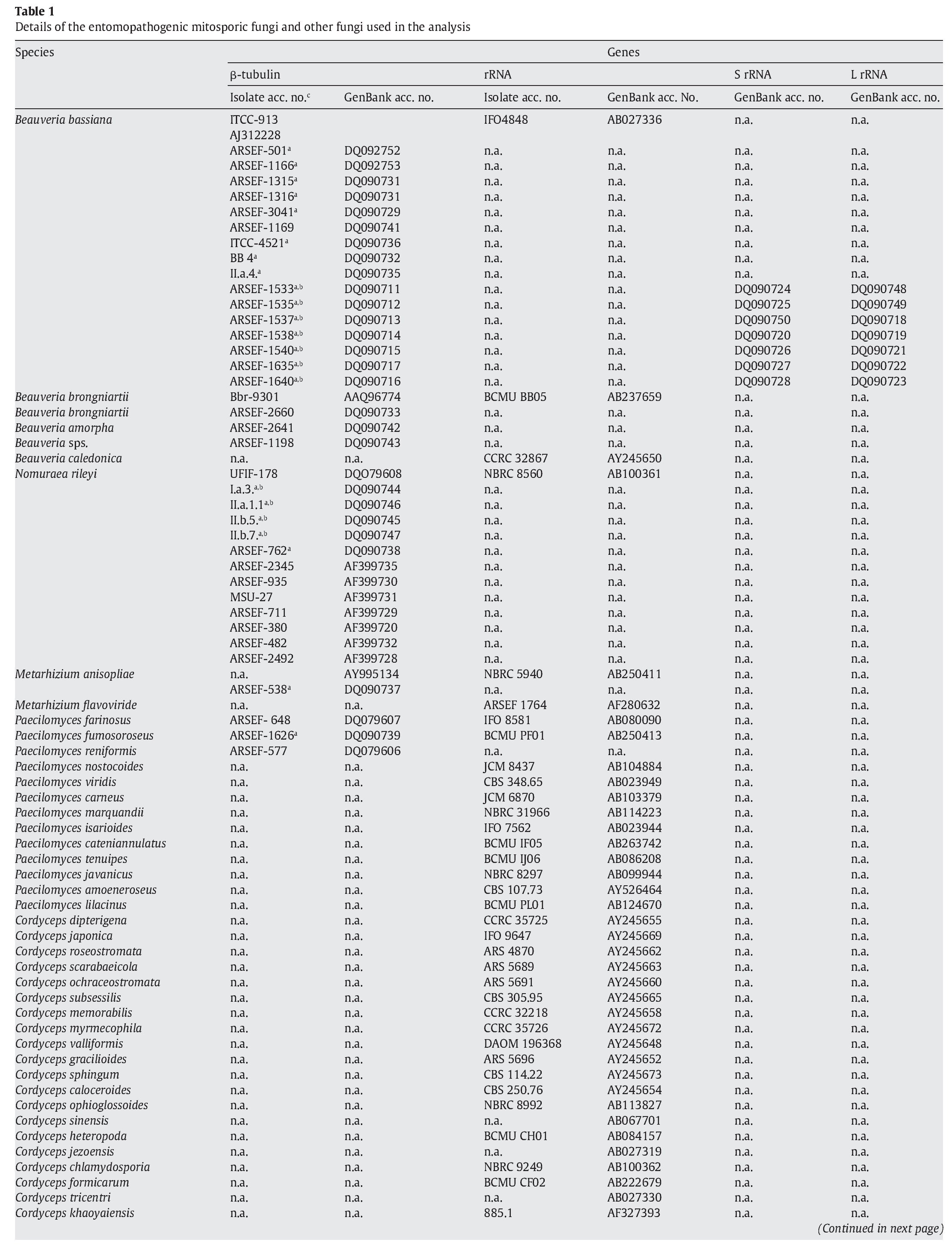
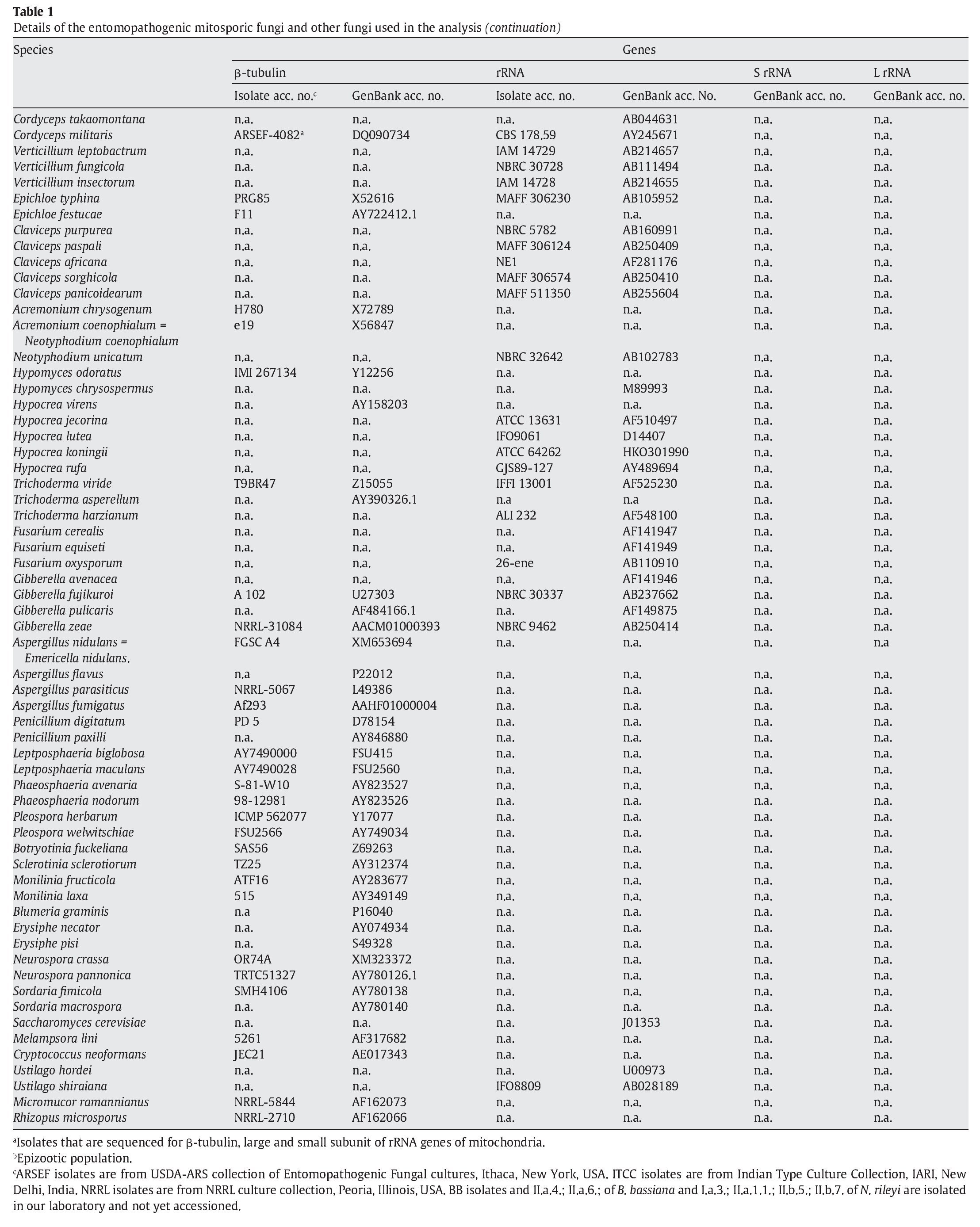
A fungal sample constituting 147 fungal entries representing 94 species, 21 of which are asexual and entomopathogenic, was analysed (Table 1). Among them, 28 isolates were sequenced for the partial region of β-tubulin gene and the rest of the sequences were obtained from GenBank database entries (http://www.ncbi.nlm.nih.gov/ Genbank/ index.html) (Table 1).
The partial regions of the three genes, namely β-tubulin, small and large subunits of mitochondrial rRNA genes, were amplified as described by Uma Devi et al35. The fungal sequences related to these sequences were retrieved from Genbank using B. bassiana sequences with accession numbers AJ312228 (β-tubulin) and AB027336 (rRNA) as query sequences in blast search with default parameters. An E-value cutoff of 2e-119 (β-tubulin) and 2e-95 (rRNA) was used to assign homology. In addition, partial nucleotide sequences of β-tubulin gene of seven N. rileyi isolates8 were included in the sample.
The sequences were initially aligned and later realigned until good alignment was obtained using the multiple alignment program AlnExplorer of MEGA ver. 3.014. Flanking regions that were not part of significant multiple alignment were trimmed off. The consistently aligned regions that have shared/derived characters of the homologous regions were used as feed for various tree construction methods. DNA sequence alignments were submitted in TreeBASE (http://www.treebase.org/treebase/), a relational database designed to manage and explore information on phylogenetic relationships with accession number SN2616.
To infer the position of asexual entomopathogenic fungi in the fungal kingdom, a data set including partial amino acid sequences of the β-tubulin gene with 44 entries (Table 1) was analysed. To infer the position of asexual entomopathogenic fungi in the order Hypocreales, a data set including partial nucleotide sequences of the rRNA gene with 66 entries (Table 1) was analysed. To examine the interrelationship among the entomopathogenic asexual fungal genera and members within a species, a data set comprising 43 partial nucleotide sequences with several isolates of the two complex species B. bassiana and N. rileyi was analysed (Table 1). To examine the cryptic speciation in the complex species B. bassiana, a data set comprising seven partial nucleotide sequences of three genes, namely β-tubulin, large and small subunit of mitochondrial rRNA genes, was analysed (Table 1).
The default parameters taken for pairwise alignment were as follows: gap opening penalty (GOP) = 10/15, gap extension penalty (GEP) = 0.1/6.6 and for multiple alignment, GOP=10/15, GEP = 0.2/6.6 with a gap separation distance = 4, transition weight of 0.5/0.5, Gonnet protein/IUB DNA weight matrix and a delay divergent cutoff % = 30/30.
The statistical procedures of minimum evolution (ME), maximum parsimony (MP) and neighbor joining (NJ) of the program MEGA ver. 3.014 were used. Distances for neighbor joining tree were calculated under the Kimura 2-parameter model. In MP and ME analyses, a single heuristic search was performed with Close Neighbor Interchange (CNI) branch swapping. For ME, NJ tree was used as the starting tree for heuristic search. For MP, stepwise addition procedure was employed. The maximum number of trees that could be saved during the heuristic search procedures was set to 1000. When multiple trees were found under ME and MP procedures, a single consensus tree was created. Confidence levels for individual branches were determined by bootstrap analysis for 1000 replicates6. To address the possible misleading effects of the data set phylogenetic relationships were then inferred using selected evolutionary model and the heuristic search option for maximum likelihood implemented in PAUP ver. 4b. We implemented Kishino-Hasegawa (KH) test13 using normal approximation (two-tailed test) to statistically evaluate tree topology against a series of topologies. In addition, trees were inferred with Bayesian inference using MrBayes, ver. 3.04b10. A Metropolis-Coupled Markov Chain Monte Carlo (MCMCMC) starting tree was initiated at random and run for 10 million generations. Trees were sampled each 1,000 cycles. Four chains were run simultaneously, of which three were heated and one was cold. Stationarity of the log likelihoods was monitored to verify convergence by 1,000,000 cycles. A consensus tree was made with multiple phylogenetic trees to determine the posterior probabilities at different nodes. To assess cryptic speciation in the sequence data of B. bassiana, the maximum parsimony analysis (PAUP ver. 4b) was done. A heuristic search analysis was run with 'tree-bisection-reconnection' (TBR) branch swapping with accelerated transformation (ACCTRAN) optimization to infer branch lengths with MULTREES option on, ADDSEQ set at random and 10 randomized replicates. All characters were weighed equally. A single tree was described when multiple trees were found.
Results
Phylogenetic position of entomopathogenic fungi in the fungal kingdom
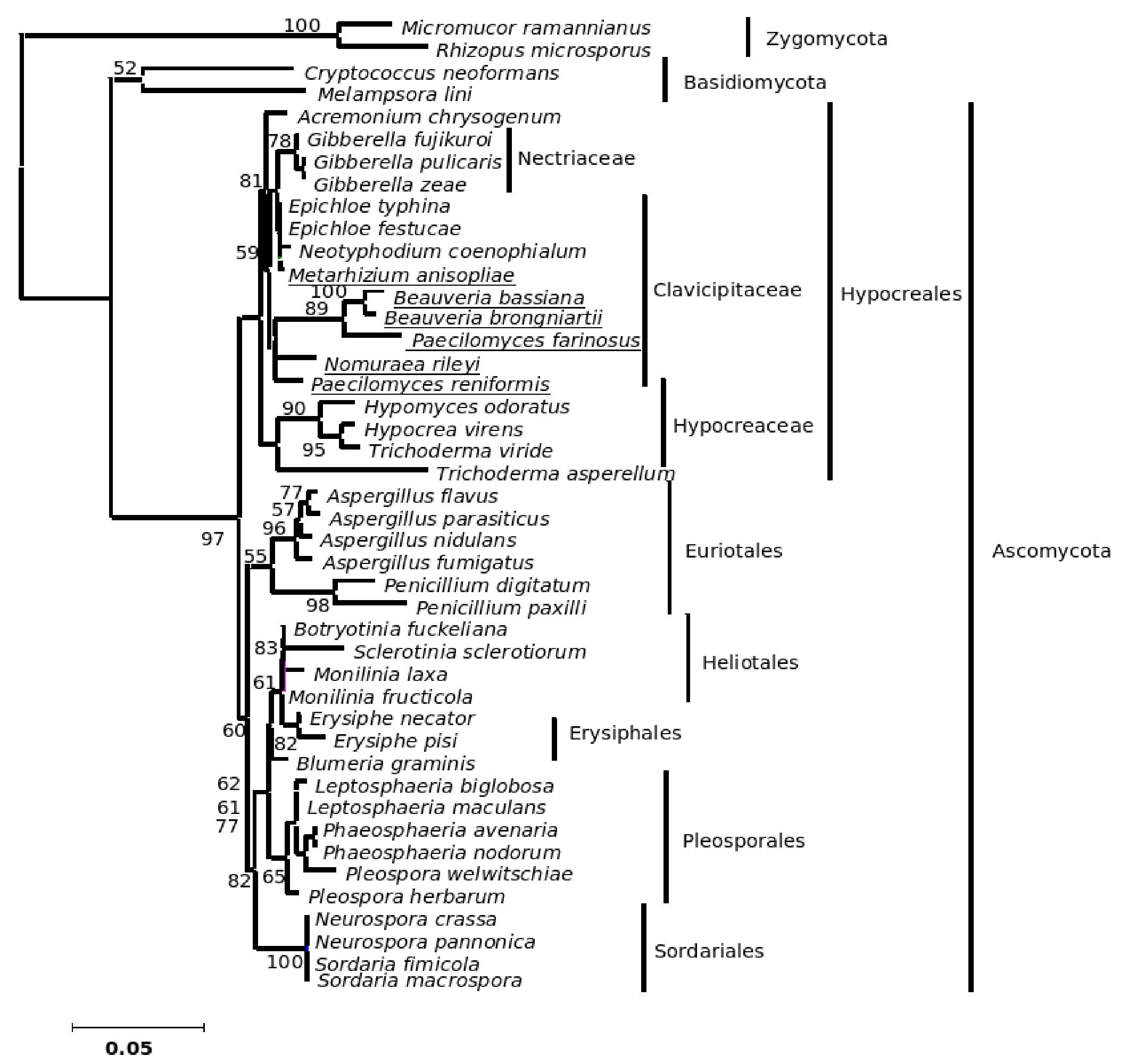
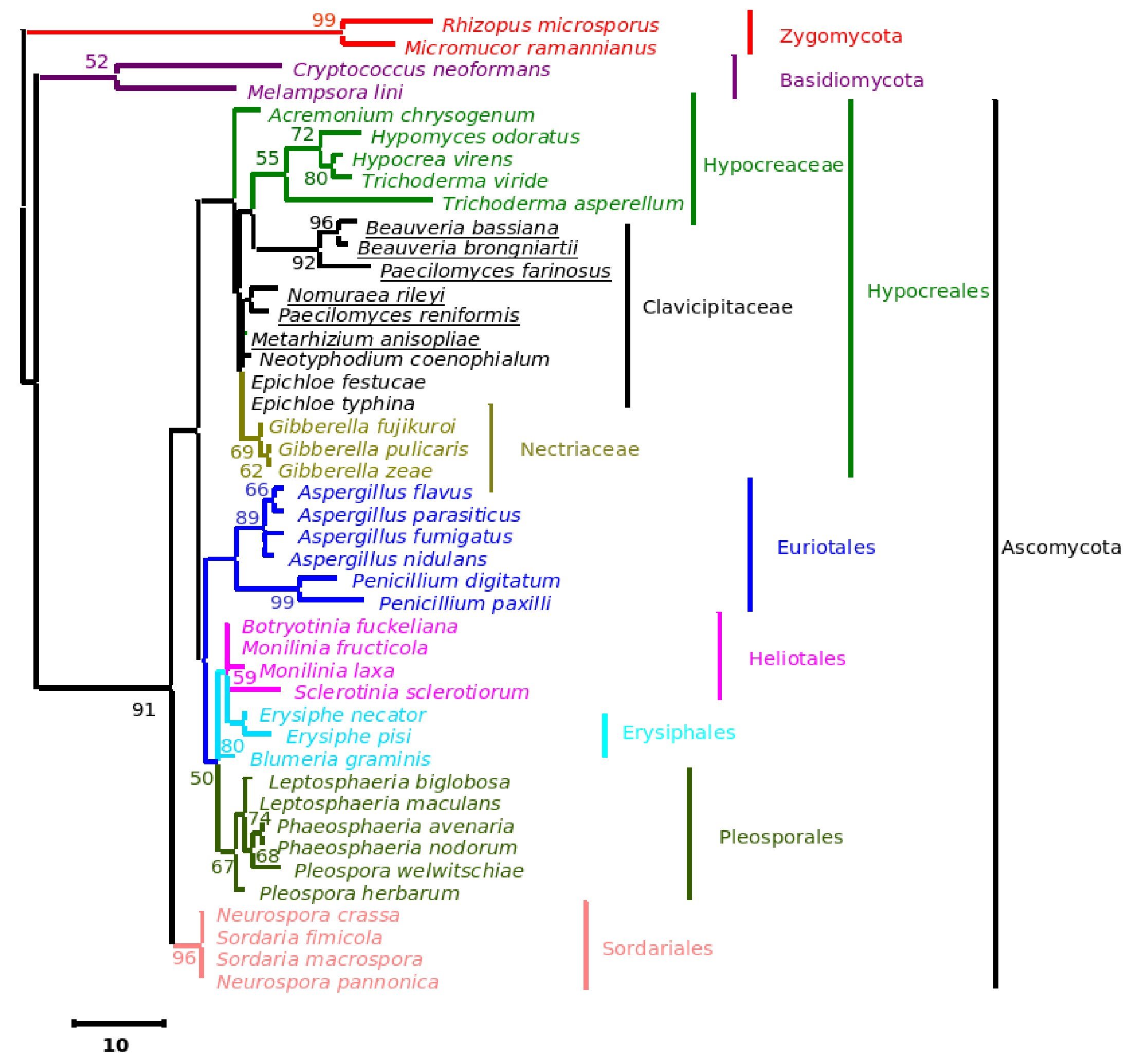
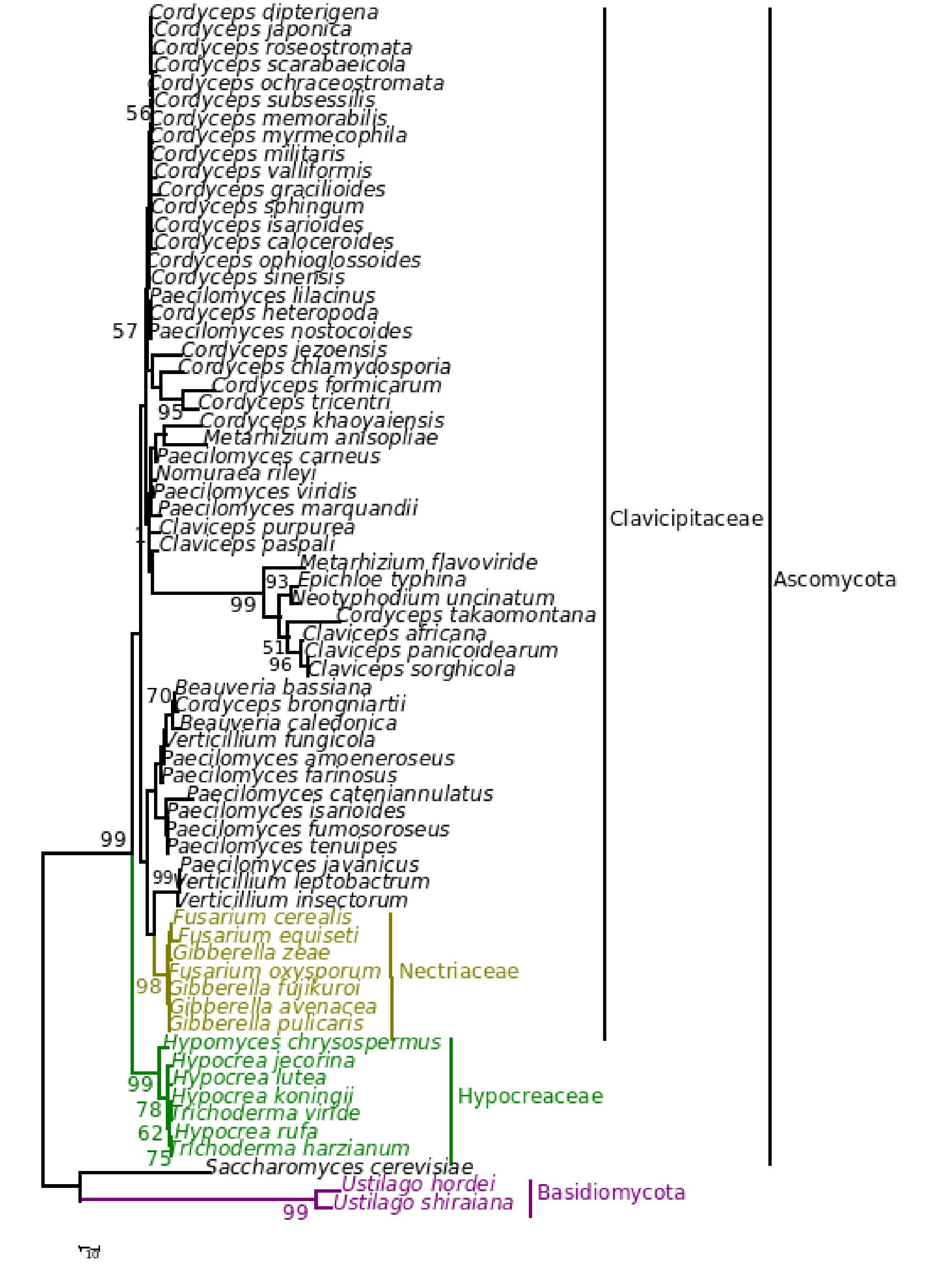
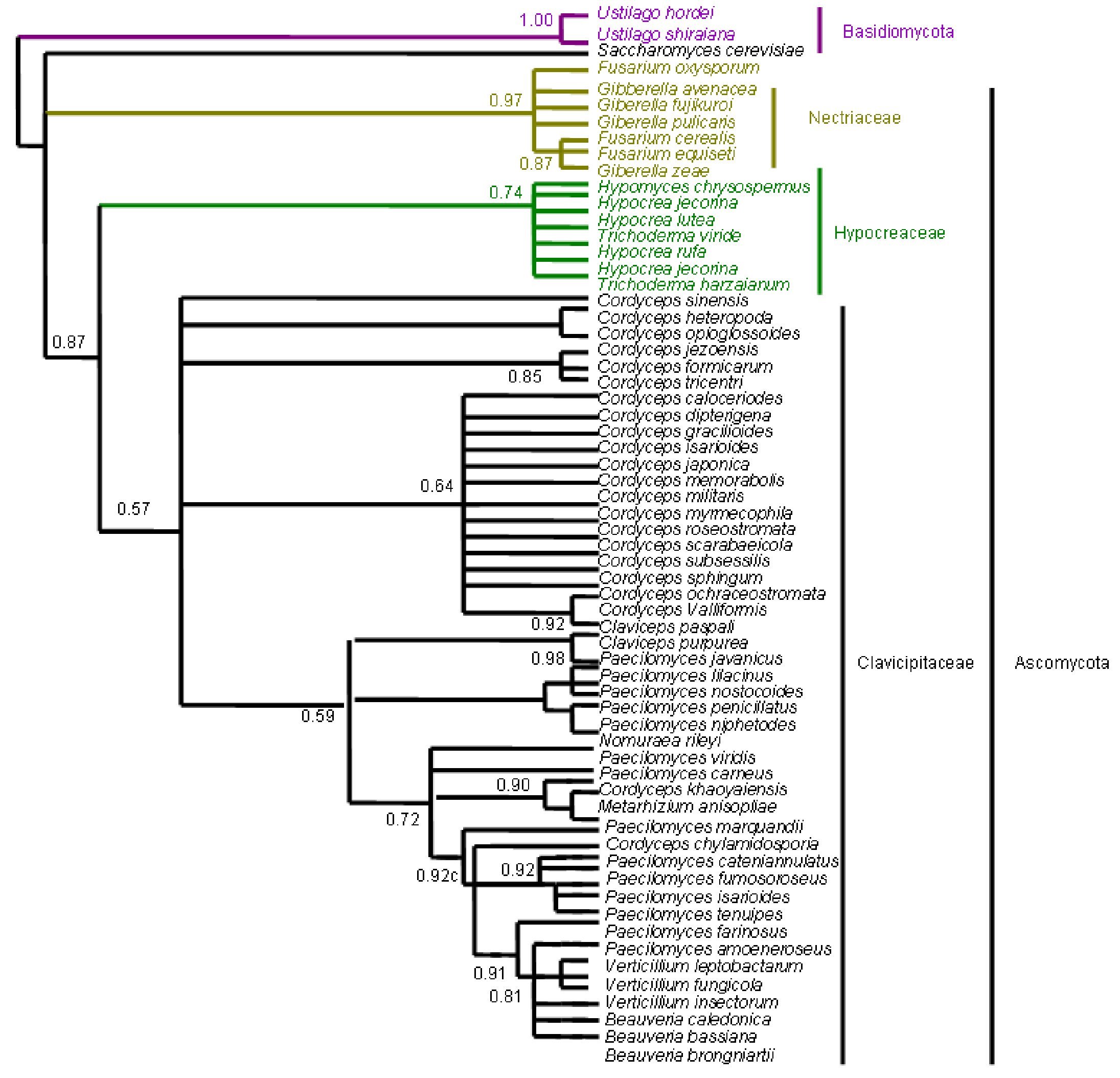
The phylogenetic trees generated based on β-tubulin sequence-aligned data set had similar topologies in both distance and parsimony methods (ME, MP and NJ) but differed in their branching order. Topological similarity of the trees obtained by different methods (MP, ME and NJ) indicates that these clusters are not incidental. The tree derived through NJ is represented in Figure 1 and is further described. The ME and MP trees are given as supplementary material (Figs. 2 and 3). Branching order reflected the expected pattern with fungi of the three divisions Asco, Basidio and Zygomycota separating into three major clades (Fig. 1). The whole clade of Ascomycetes was well supported with a bootstrap value of 91%. The Ascomycota clade branched into two sub-clades with the separation of members of Hypocreales in one group and the rest in the other (Fig. 1). The sub-clade with non-hypocrealean fungi fissured into five branches each representing members of one taxonomic order - the Sordariales, Heliotales, Erysiphales, Pleosporales and Euriotales. There were three sub-clades in the hypocrealean clade each representing one family: Nectriaceae, Hypocreaceae and Clavicipitaceae (Fig. 1). All the mitosporic entomopathogenic fungi B. bassiana,Beauveria brongniartii, N. rileyi, Paecilomyces fumosoroseus, Paecilomyces farinosus and Metarhizium anisopliae grouped into the claviciptalean sub-clade of Hypocreales (Fig. 1). Within this sub-clade, each of the entomopathogenic fungal genus separated into a different clade (Fig. 1). M. anisopliae was found to be closely related to sexual clavicipitalean fungi (Fig. 1). There was one discrepancy in the distribution of the members of Hypocreaceae. Sexual Ascomycete, Acremonium chrysogenum of Hypocreaceae, separated from the other members of that family (Fig. 1). The Bayesian tree of highest likelihood (Fig. 4, supplementary material) has a significant (>95%) posterior probability and strong bootstrap support for all (ME, MP and NJ) trees. p value in the KH test (p = 0.05) indicates that the maximum likelihood tree (5154.26590) is not significantly worse than the original maximum likelihood tree (5155.47481).
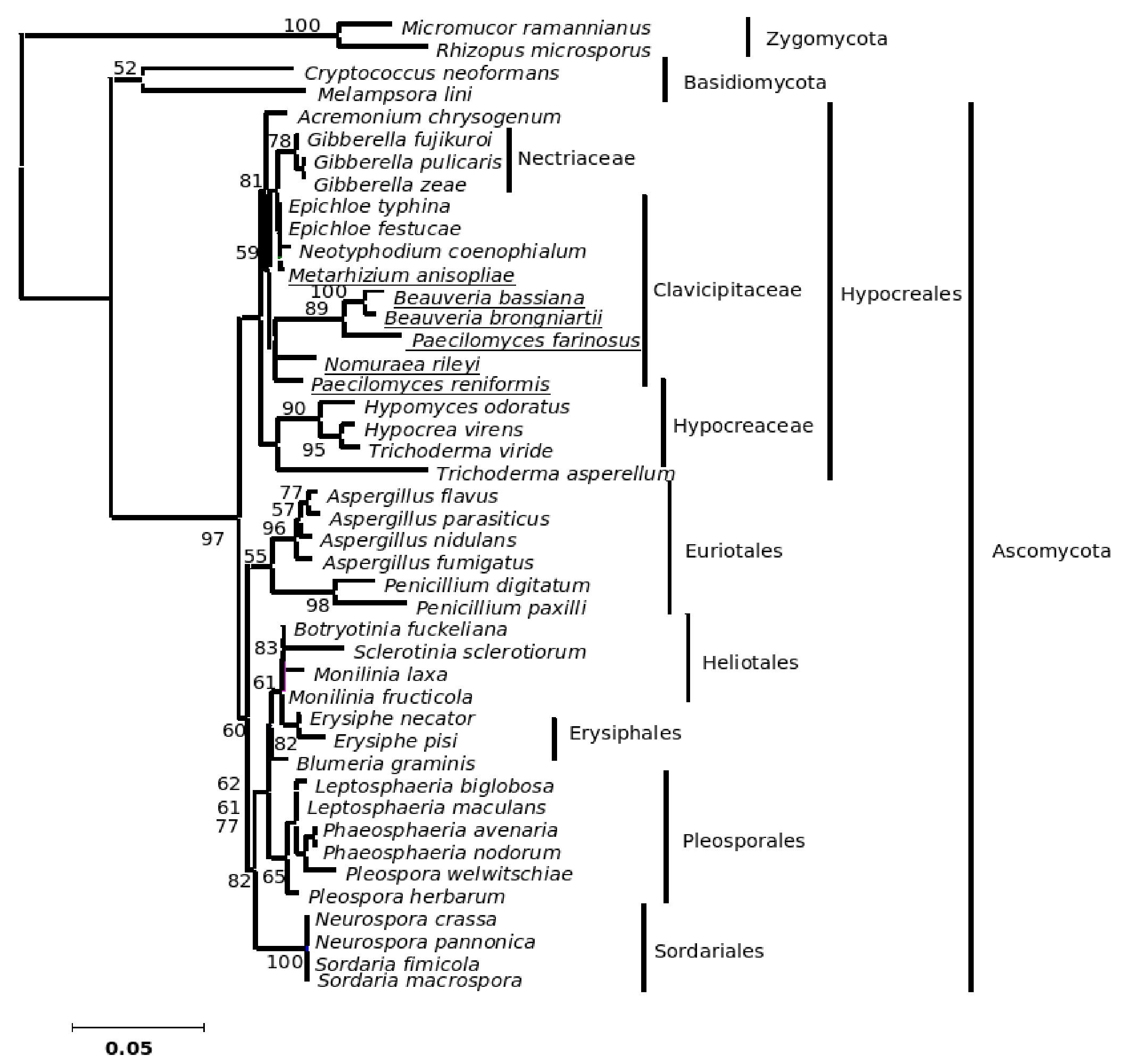
Figure 1. Consensus phylogenetic tree derived by neighbor joining method of a sample of 44 fungal entries to assess the phylogenetic affiliation of the asexual entomopathogenic fungi Beauveria bassiana and Nomuraea rileyi to each other and other asexual fungi with entomopathogenic habit and sexual (meiosporic) fungi. Taxa underlined are entomopathogenic fungi of interest. Bootstrap values (based on 1,000 replicates) when above 50% are indicated on the branches. The tree was derived from partial sequence (271 amino acids) of the β-tubulin gene of which 150 were conserved, 121 were variable, of which 83 were parsimony informative.
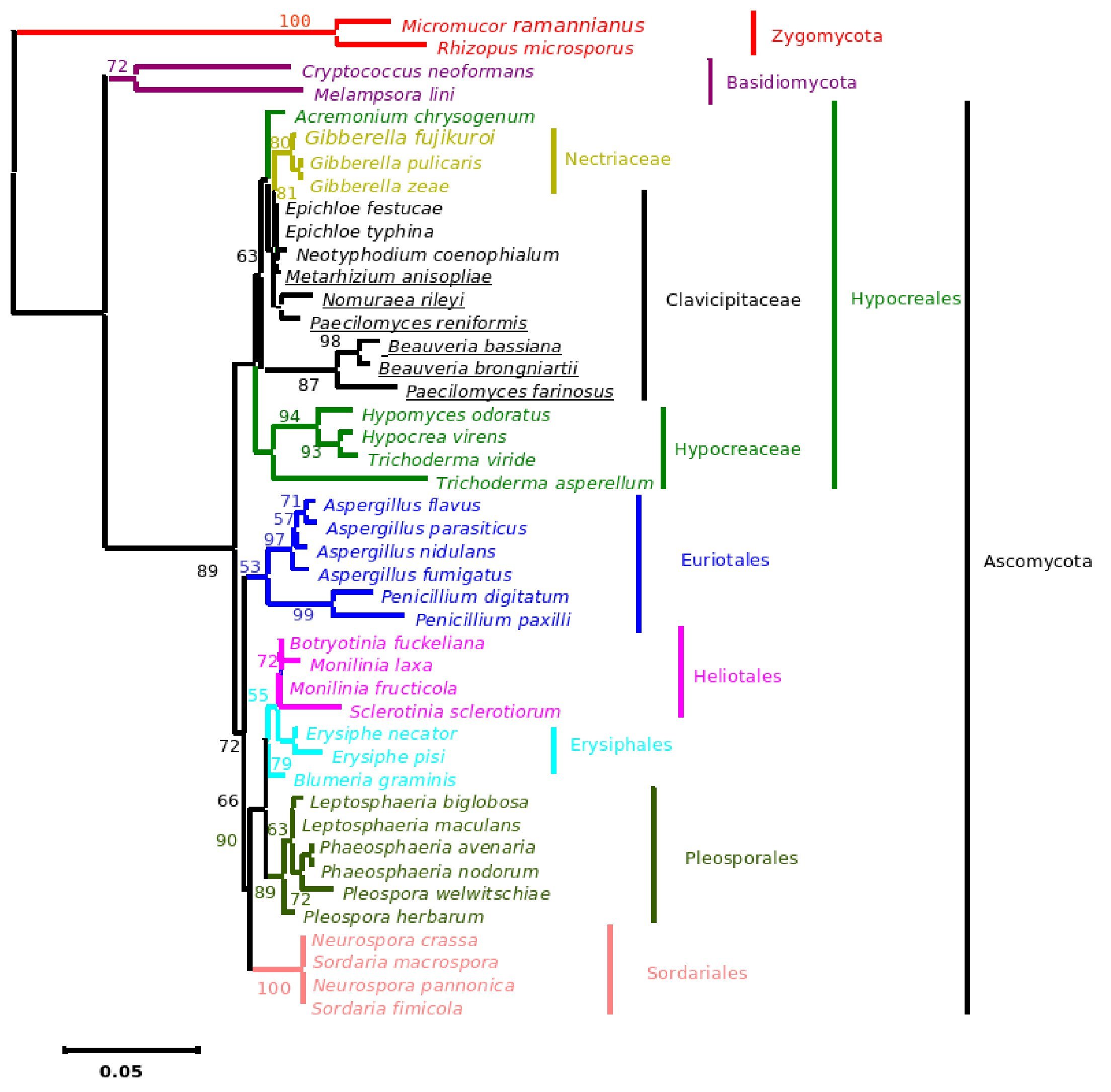
Figure 2. Consensus phylogenetic tree derived for the same data set as in Figure 1 derived by the minimum evolution method.
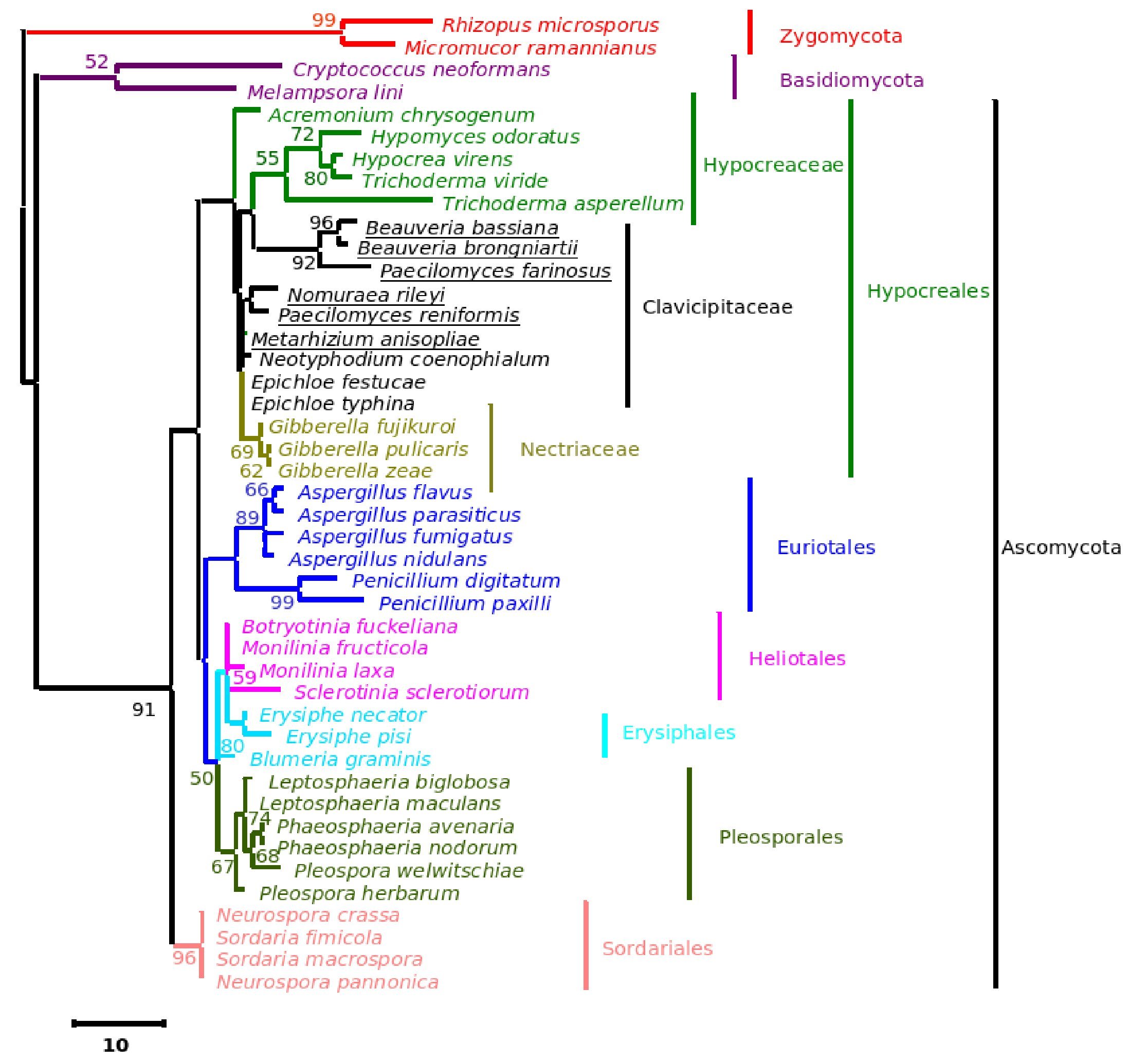
Figure 3. Consensus phylogenetic tree derived for the same data set as in Figure 1 derived by maximum parsimony analysis. Component tree statistics: tree length (L) = 269; consistency index (CI) = 0.737; retention index (RI) = 0.818.
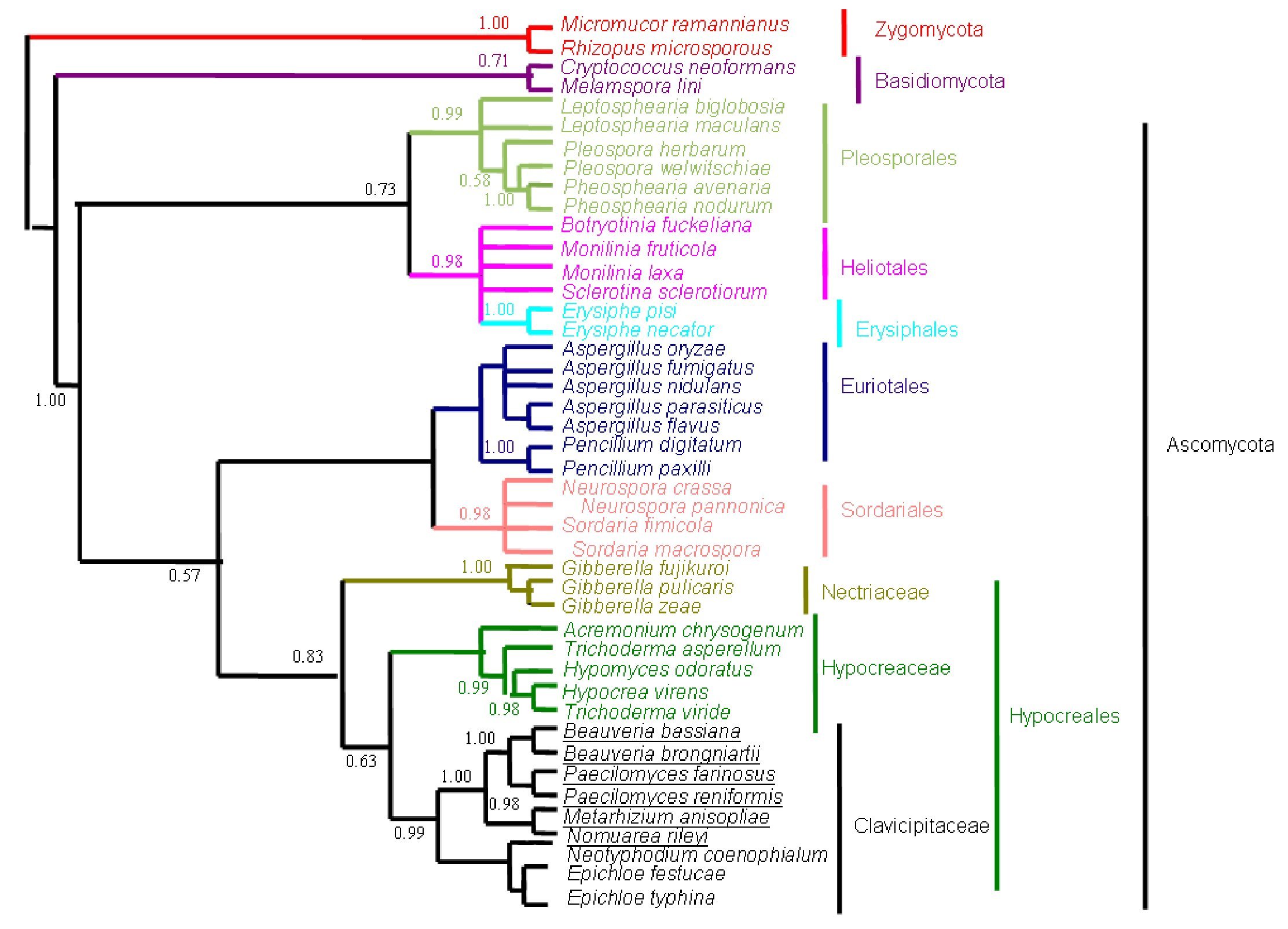
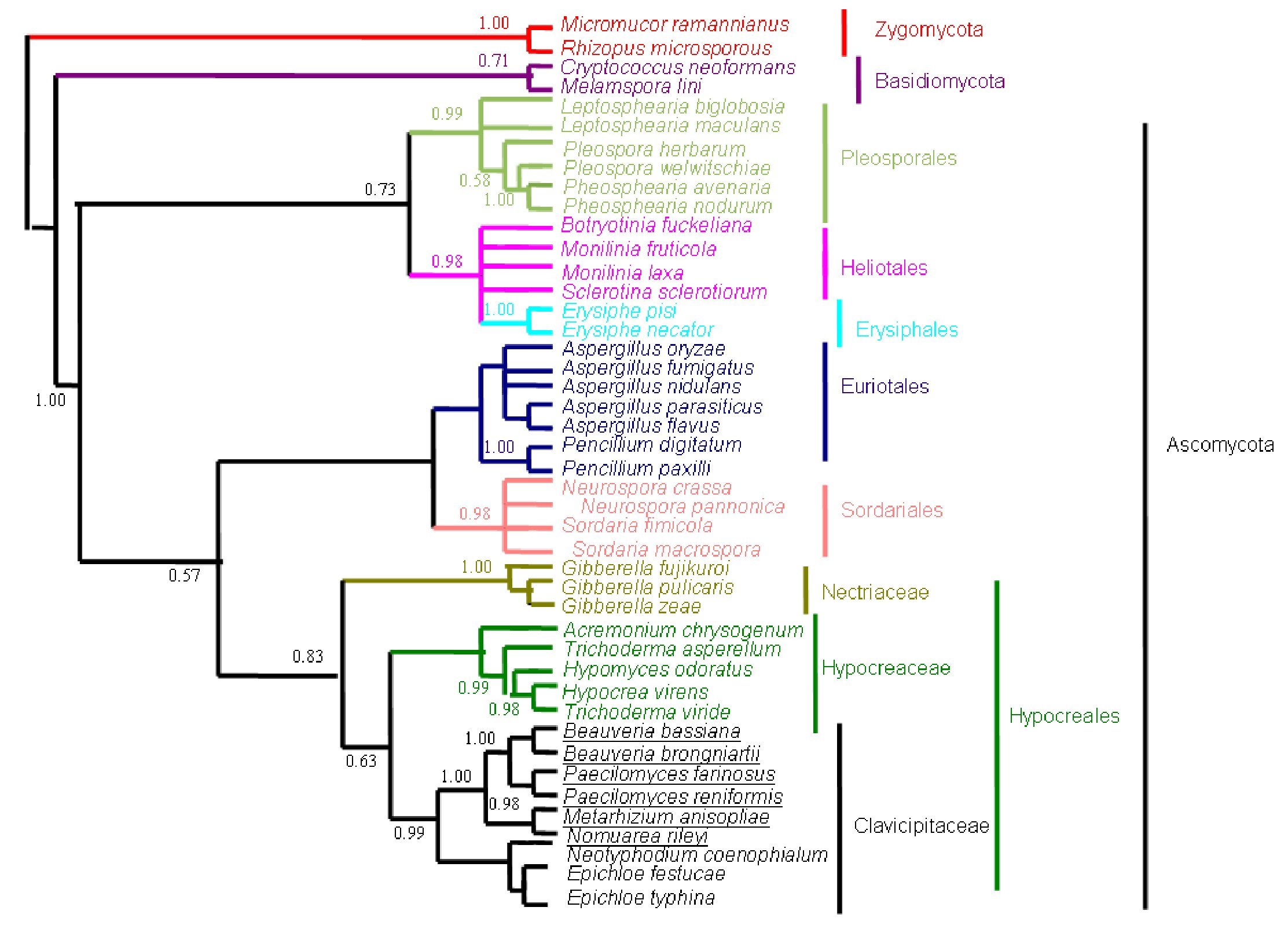
Figure 4. Bayesian Metropolis-coupled Markov chain Monte Carlo (MCMCMC) fungal tree depicting phylogenetic relationships for the same data set as in Figure 1. This phylogeny resulted from a 50% majority rule consensus of 10001 trees sampled with Bayesian MCMCMC. The resulting posterior probabilities (PP) are shown above internal branches. Taxa underlined are entomopathogenic fungi of interest. Species names are colored according to their respective phyla.
The results from the analysis of rRNA gene sequence data (with some differences in the fungal entries included) very closely matched the phylogenetic relationships evident in the analysis of the sequence data of β-tubulin gene. The NJ tree from the rRNA gene is given in Figure 5 (the ME, MP and Bayesian trees are provided as supplementary Figs. 6, 7 and 8, respectively).
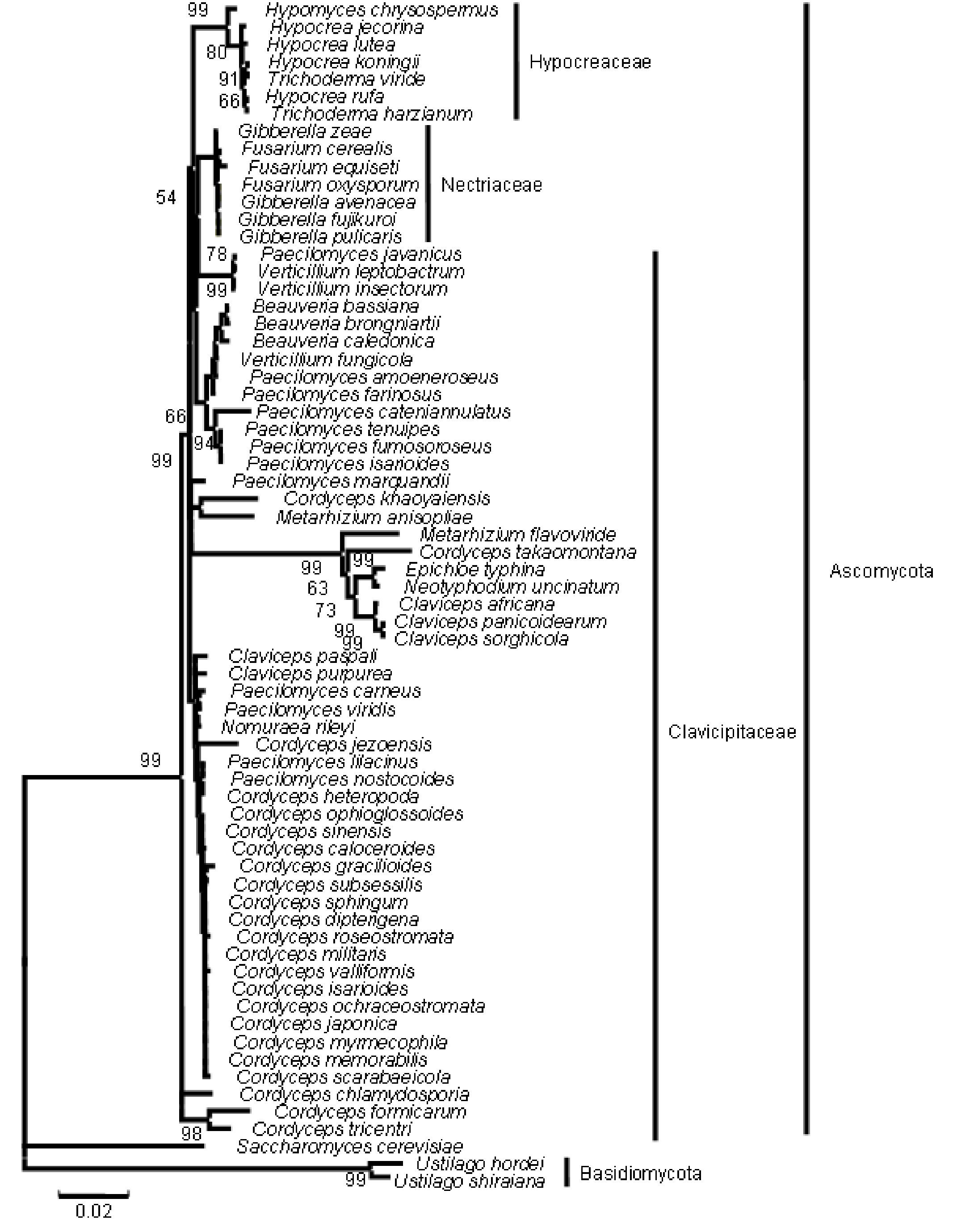
Figure 5. Consensus phylogenetic tree of a sample of 64 fungal entries to assess the phylogenetic affiliation of the asexual entomopathogenic fungi Beauveria bassiana and Nomuraea rileyi to each other and other asexual fungi with entomopathogenic habit and sexual (meiosporic) fungi derived by neighbor joining method. The tree was derived from 64 taxa and partial sequence (1359 nucleotides) of the rRNA gene of which 933 were conserved, 408 were variable, of which 293 were parsimony informative.
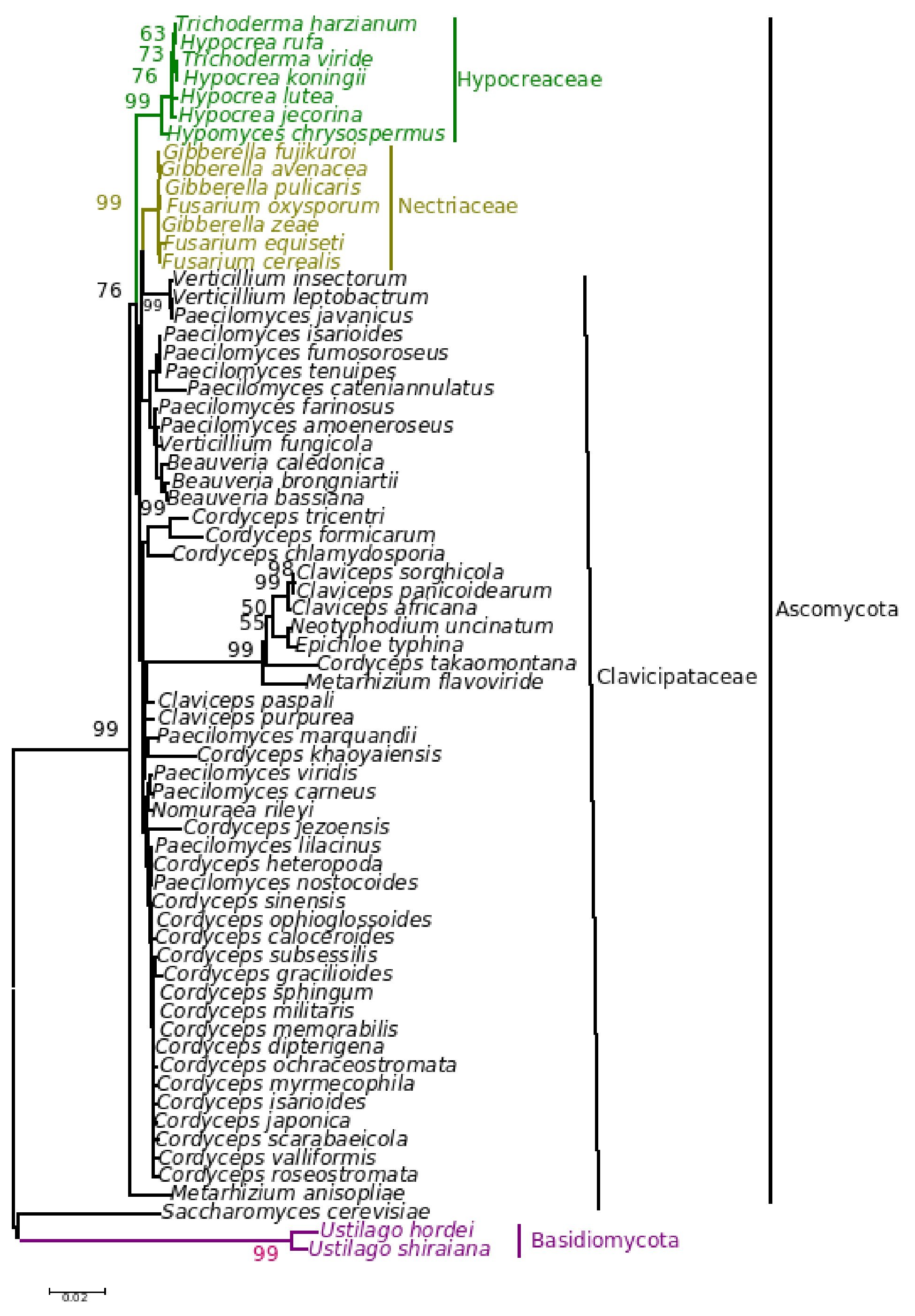
Figure 6. Consensus phylogenetic tree derived for the same data set as in Figure 5 derived by the minimum evolution method.
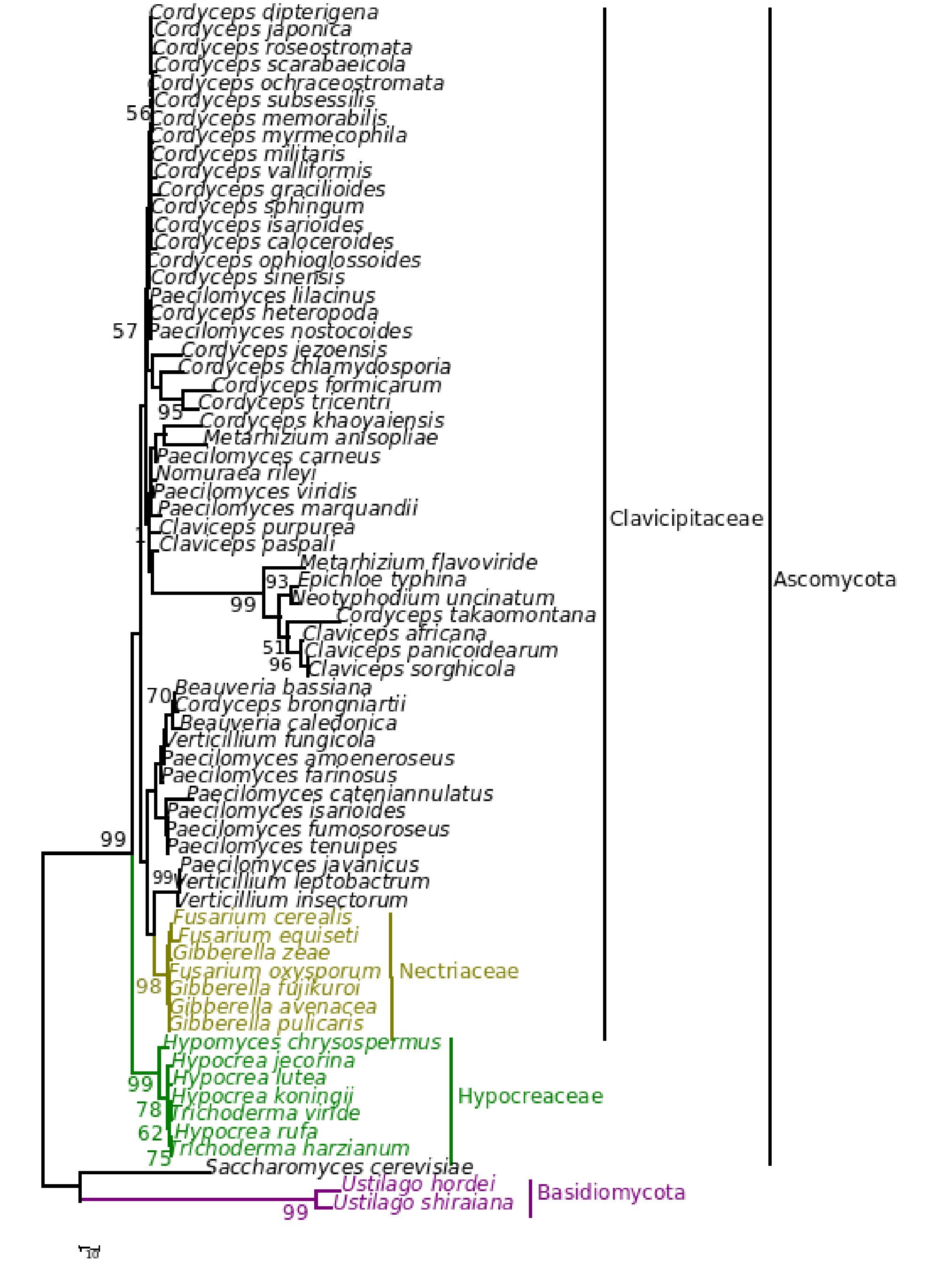
Figure 7. Consensus phylogenetic tree derived for the same data set as in Figure 1 derived by maximum parsimony analysis. Component tree statistics: tree length (L) = 574; consistency index (CI) = 0. 0.721; retention index (RI) = 0.818.
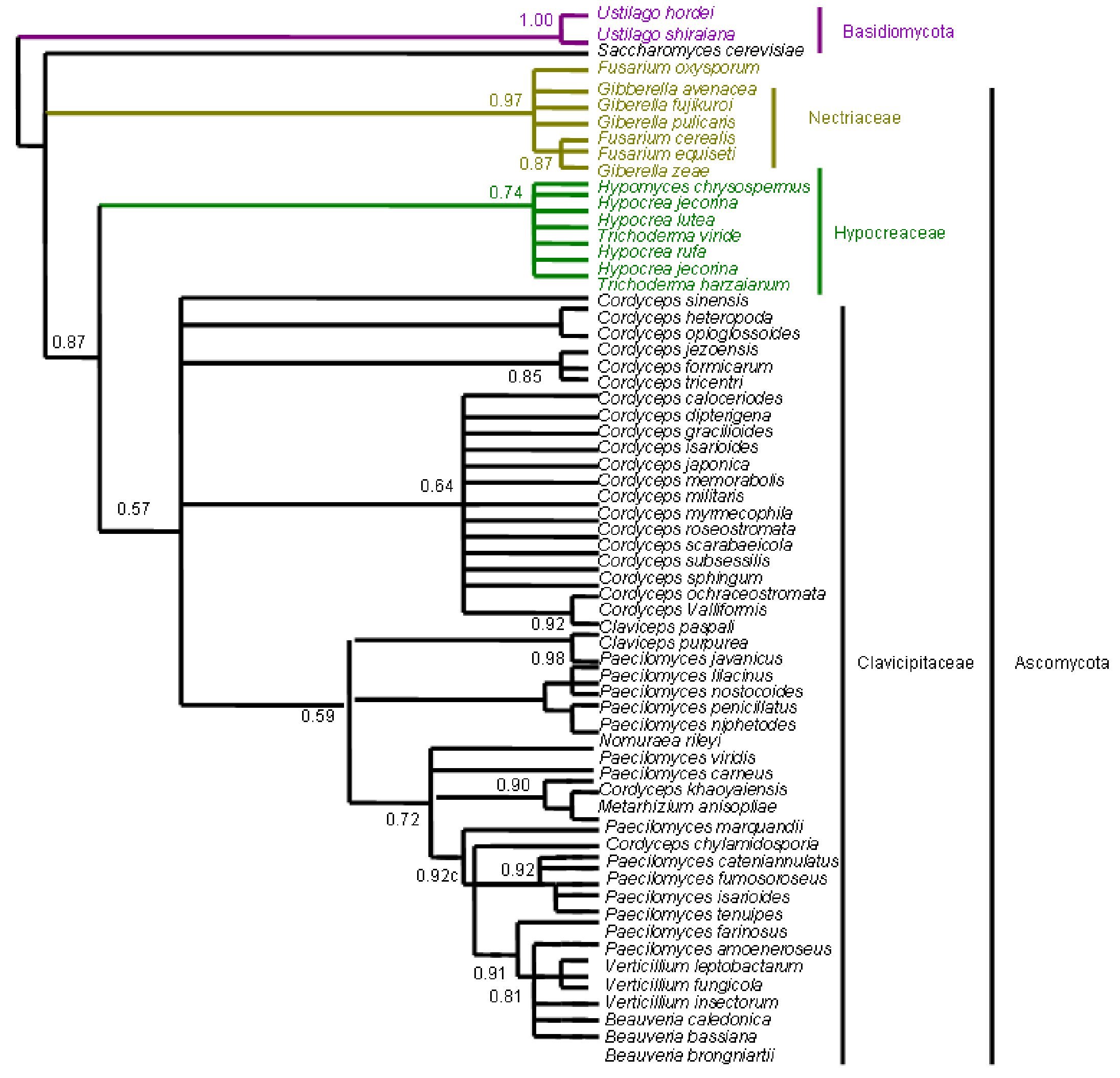
Figure 8. Bayesian Metropolis-coupled Markov chain Monte Carlo (MCMCMC) fungal tree depicting phylogenetic relationships for the same data set as in Figure 5. This phylogeny resulted from a 50% majority rule consensus of 10001 trees sampled with Bayesian MCMCMC. The resulting posterior probabilities (PP) are shown above internal branches. Taxa underlined are entomopathogenic fungi of interest. Species names are colored according to their respective phyla.
With the analysis of both β-tubulin and rRNA gene sequences, enough evidence was not found to reject the monophyly of the entomopathogenic fungal group studied. We therefore conclude that the mitosporic entomopathogenic fungi are members of Clavicipitaceae family.
Interrelationships between entomopathogenic fungal species
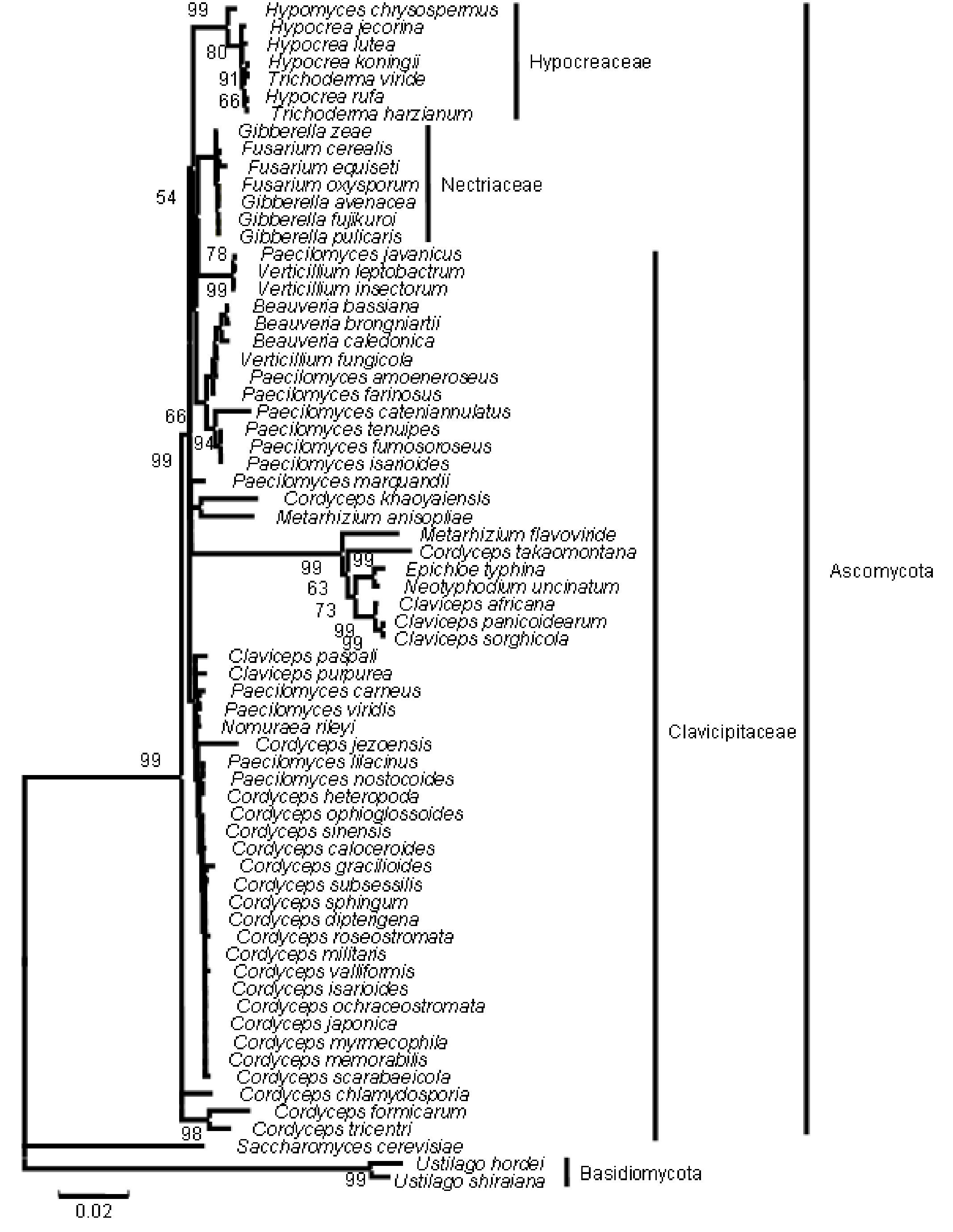
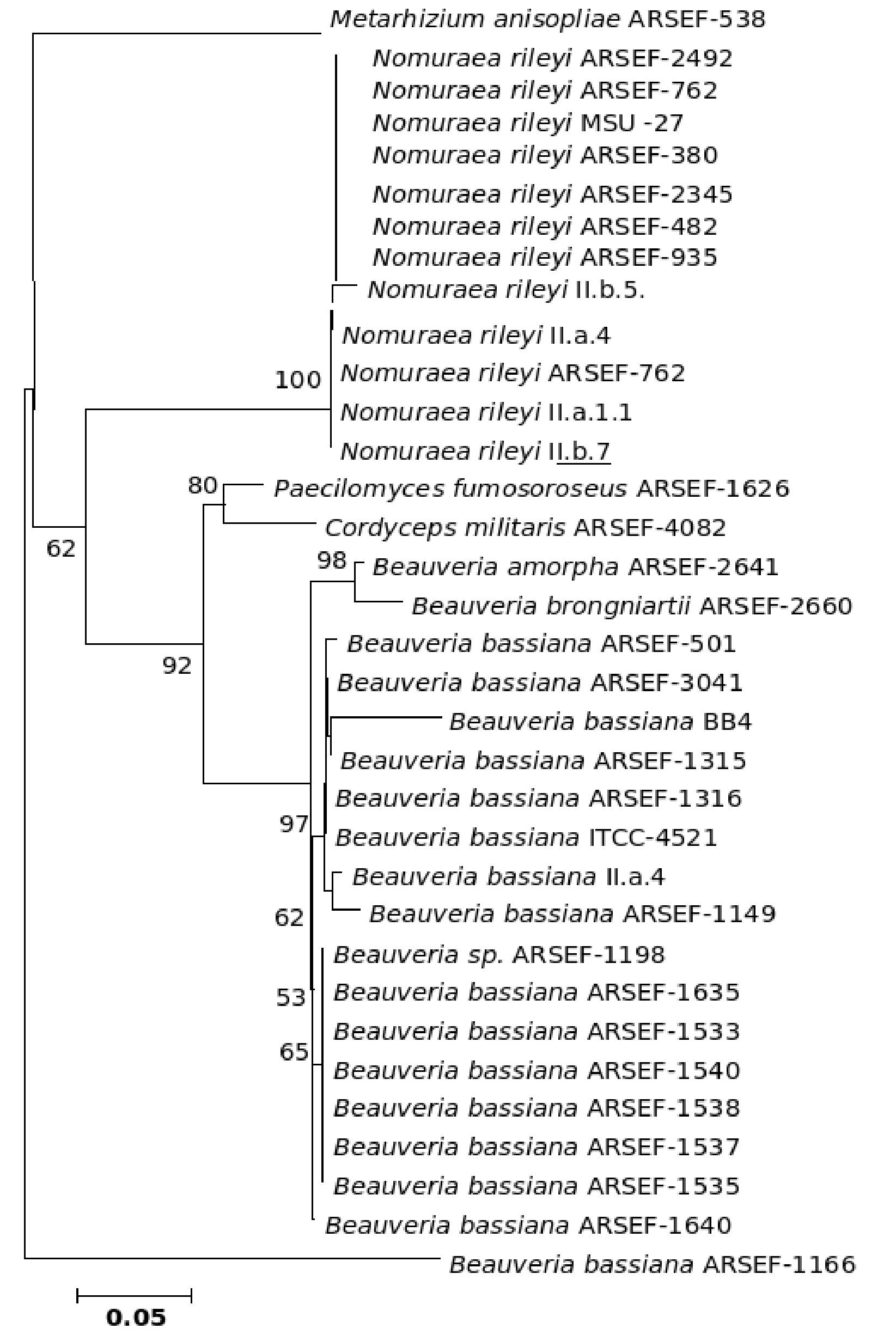
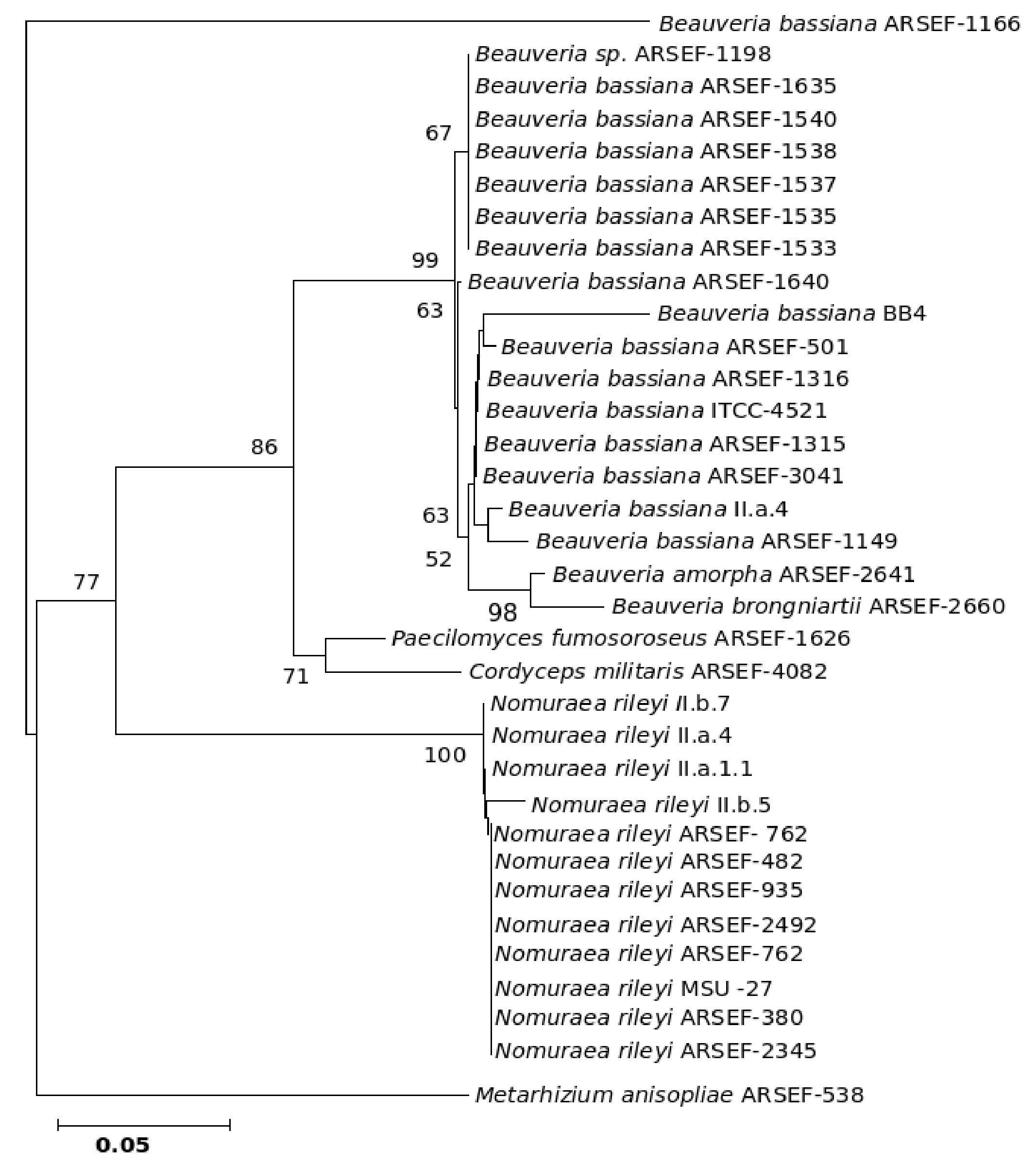
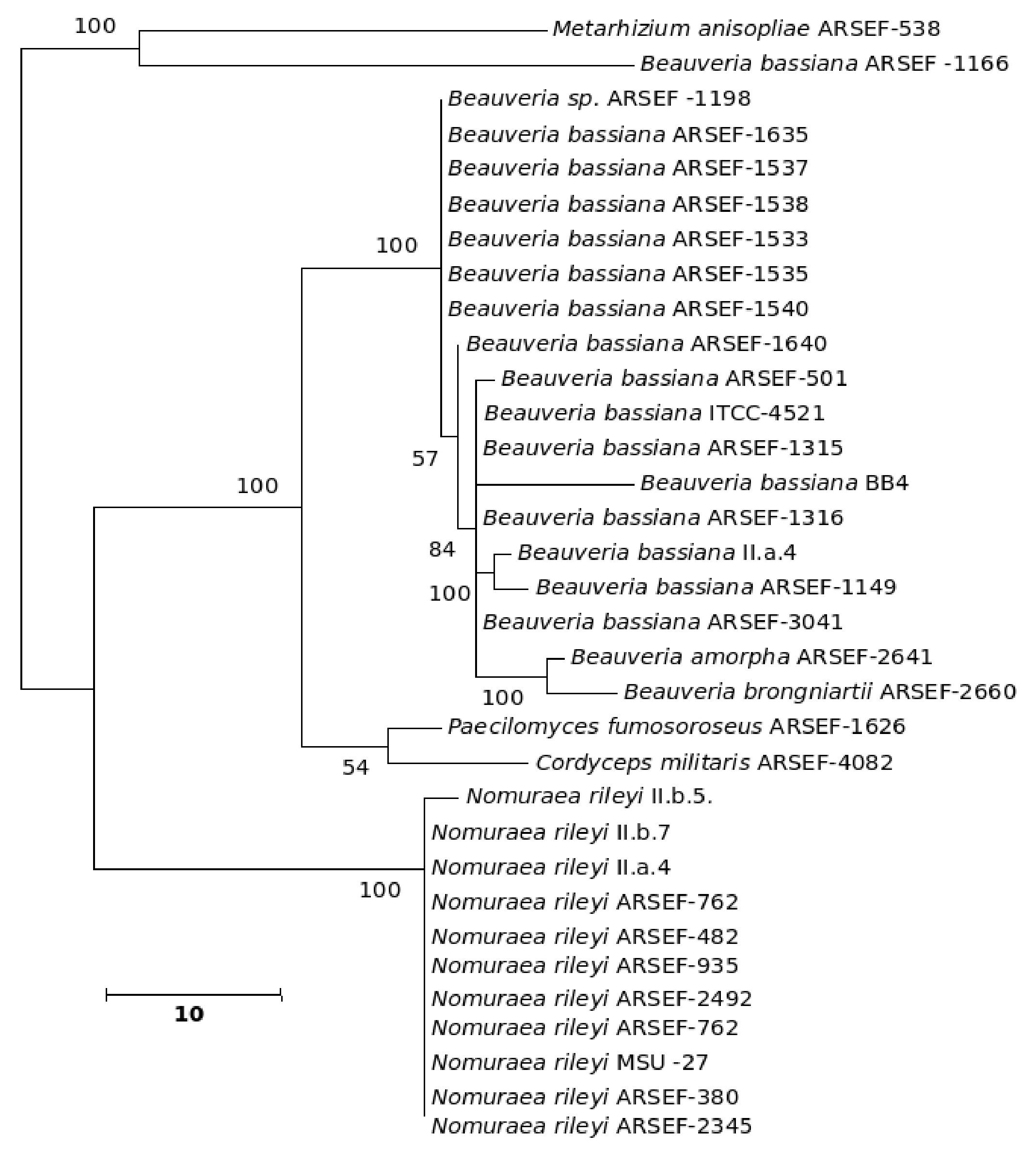
The five major genera included in the analysis separated into different clades in the trees derived by all the three (NJ, ME and MP) methods; only the position of M. anisopliae differed in the three phylogenetic trees. The NJ tree is described and is given in Figure 9; the ME and MP trees are provided as supplementary material in Figures 10 and 11. Two isolates of B. bassiana separated from the rest of the B. bassiana isolates as another clade (Fig. 9). The two other species of Beauveria in the sample - Beauveria amorpha and B. brongniartii - grouped with isolates of B. bassiana (Fig. 9). Deep phylogenetic lineages were observed in the B. bassiana clade (Fig. 9). Unlike B. bassiana, extensive phylogenetic speciation was not observed in the N. rileyi sample (Fig. 9). Only one isolate diverged into a different lineage among the members (Fig. 9). M. anisopliae separated as a different lineage in the main group (Fig. 9), while in the MP tree it grouped with the two B. bassiana isolates, which diverged from the main group; in the ME tree it separated out as a different lineage from the main group.
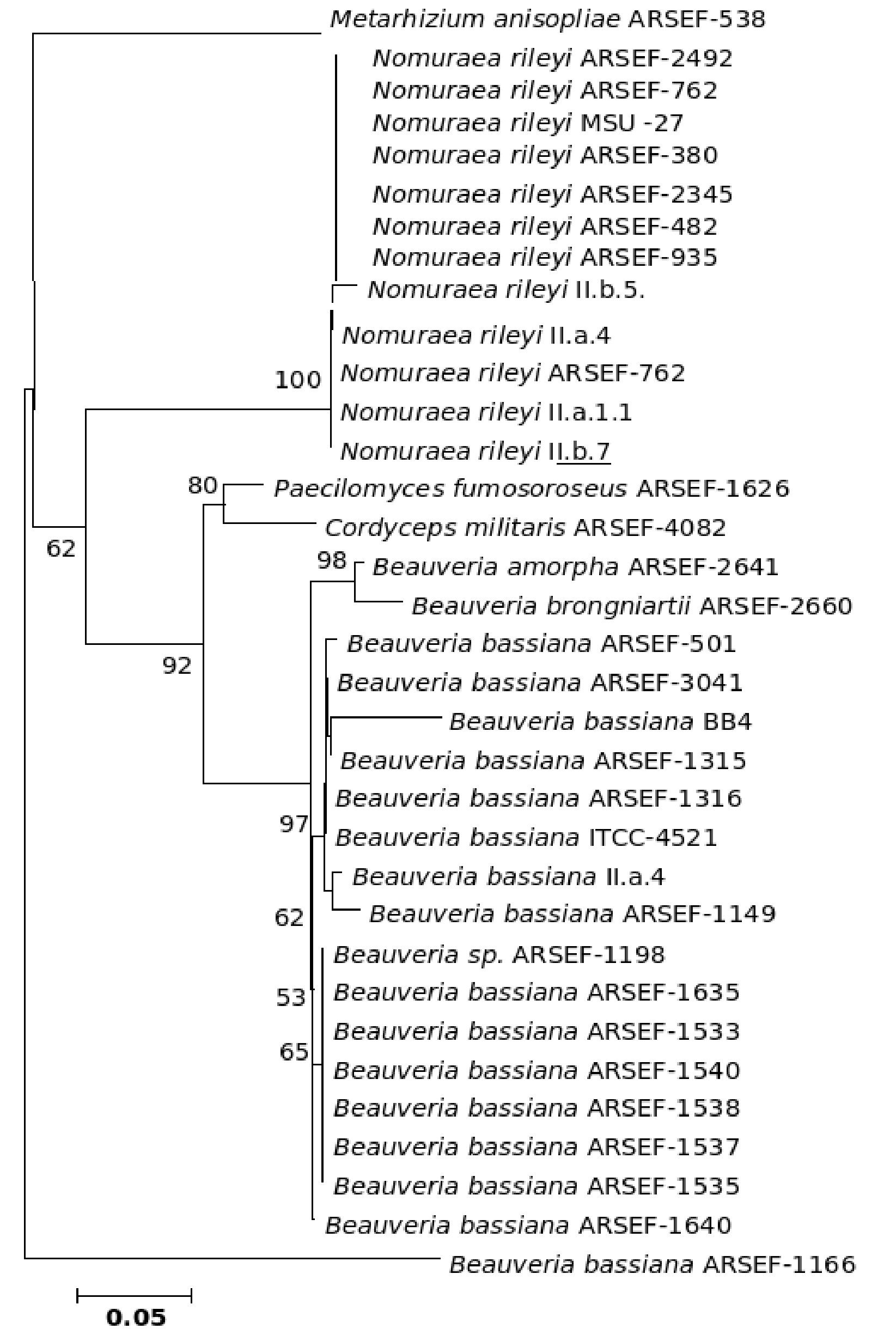
Figure 9. Consensus phylogenetic tree generated from partial β-tubulin gene sequence through neighbor joining method of a sample of isolates of Beauveria bassiana, Beauveria amorpha, Beauveria brongniartii, Nomuraea rileyi, Paecilomyces fumosoroseus, Metarhizium anisopliae, Cordyceps militaris to study the interrelationship by. The aligned data consisted of 257 characters of which 125 were conserved and the rest were variable. Of these, 82 were parsimony informative characters.
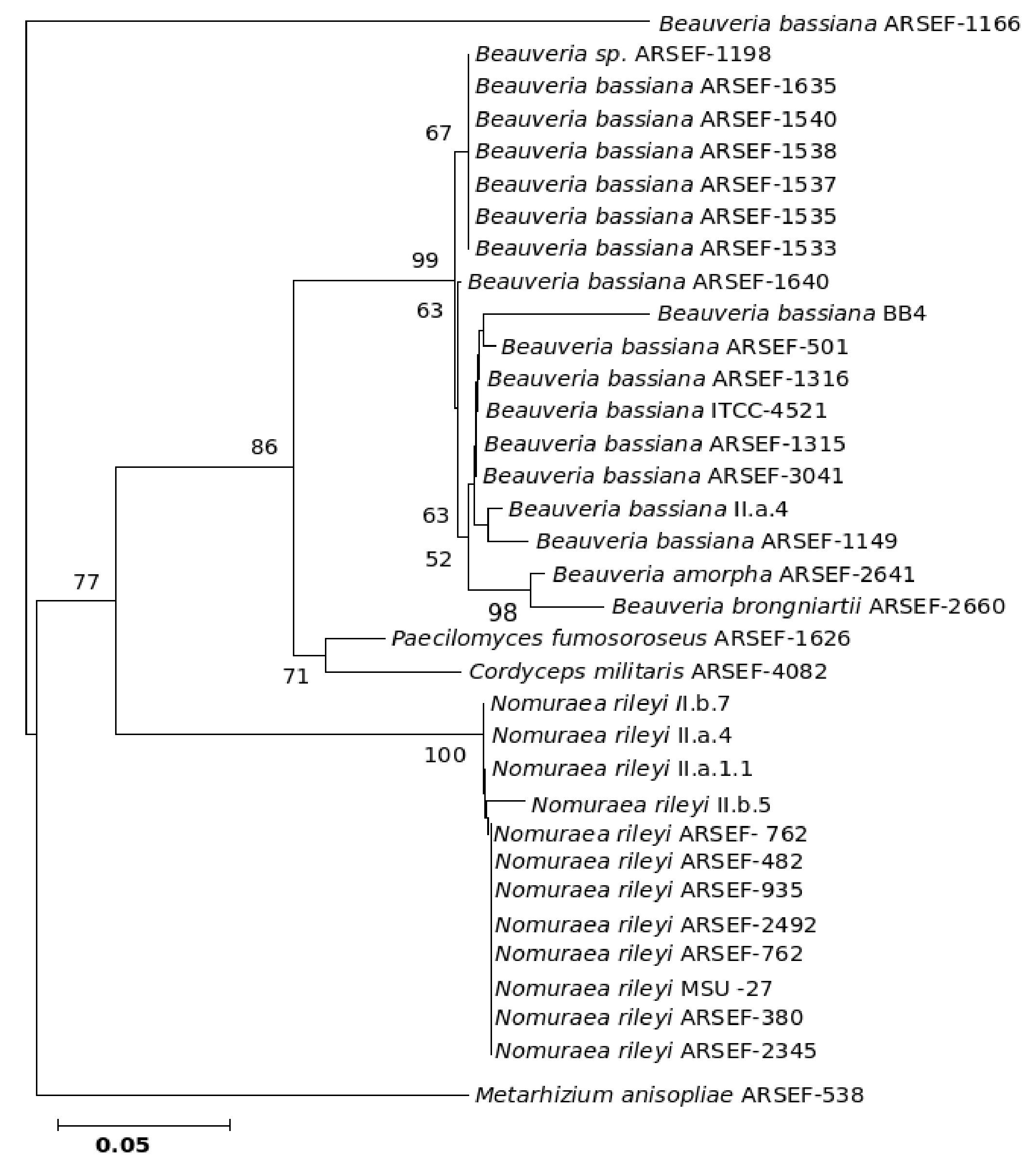
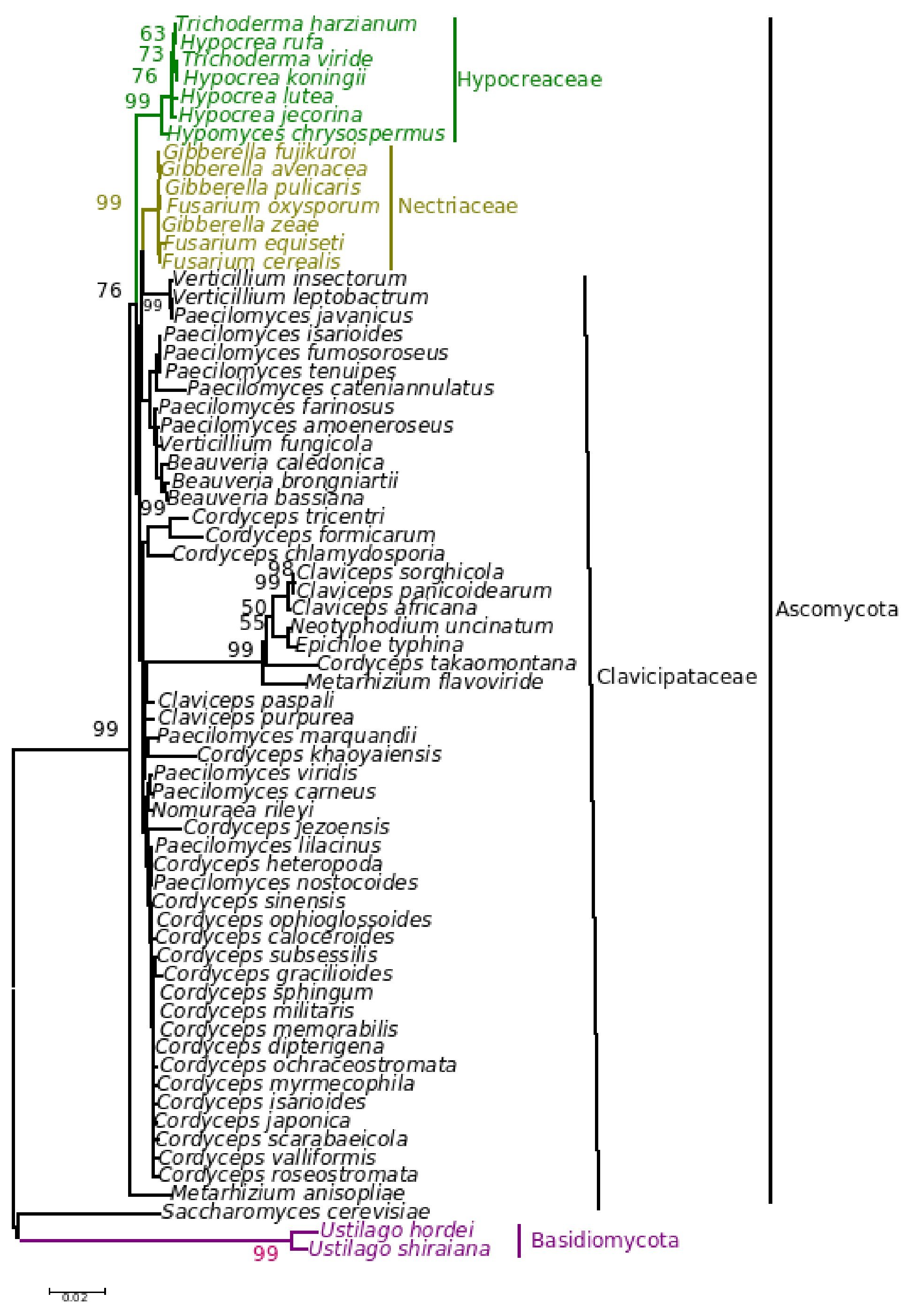
Figure 10. Consensus phylogenetic tree derived for the same data as in Figure 9 derived by the minimum evolution method. Bootstrap values when above 50% are indicated on the branches.
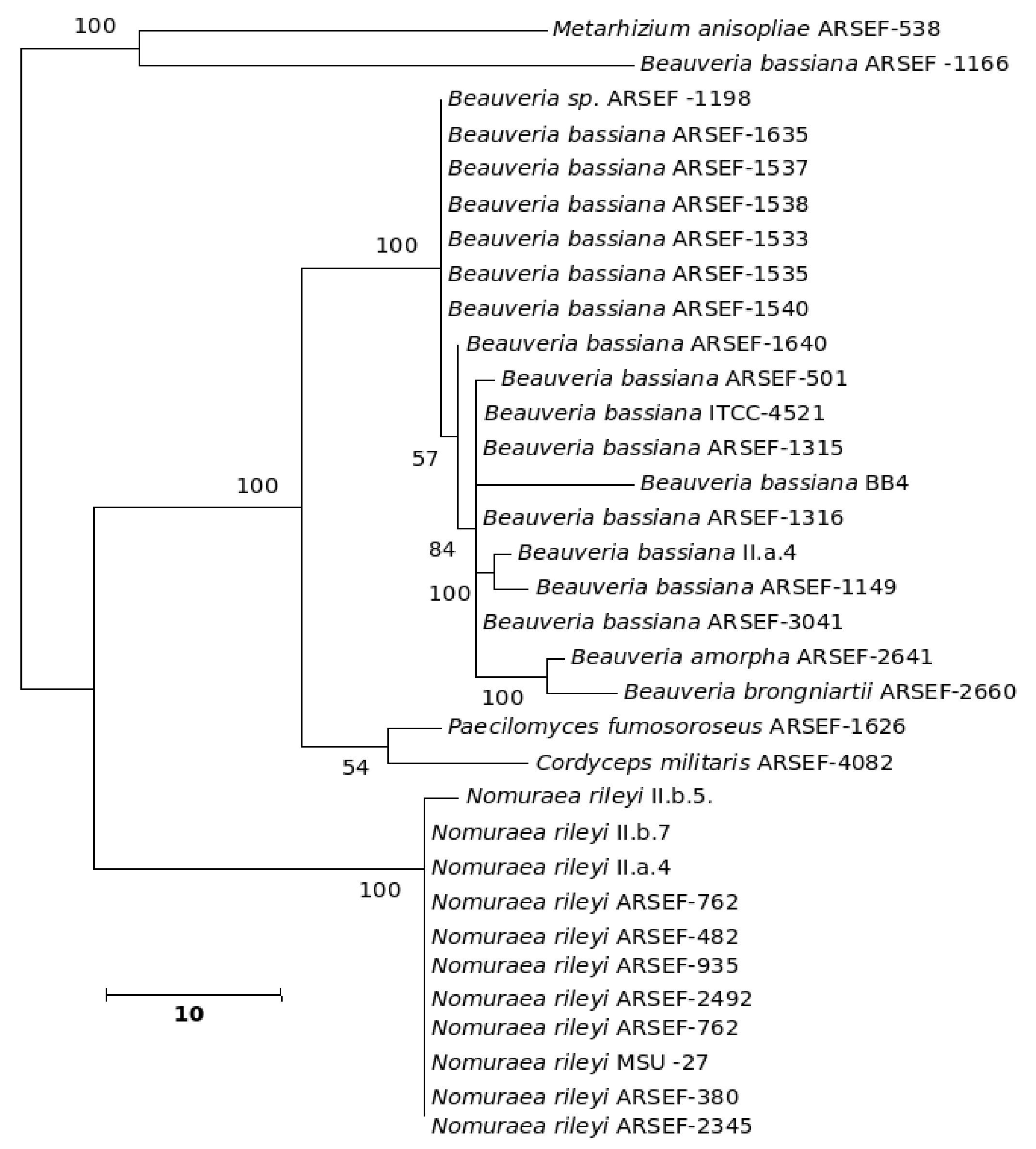
Figure 11. Consensus phylogenetic tree derived for the same data as in Figure 9 derived by the maximum parsimony method. Bootstrap values when above 50% are indicated on the branches. The description of the tree is as follows: consistency index (CI) = 0.905; retention index (RI) = 0.875 for the 326 parsimonious trees.
Cryptic speciation in Beauveria bassiana
Diagnosing species borders from single-gene phylogenies can be difficult because usually the markers do not indicate a clear cutoff between species and higher taxonomic groupings. Therefore an approach termed genealogical concordance phylogenetic species recognition (GCPSR) was used24. In fungal taxa, where mating studies are difficult to conduct and when patterns are not detectable from multilocus sequence data, multiple gene genealogies (3-10 genes) were used to uncover cryptic speciation using this powerful approach of GCPSR24. GCPSR detects genetically isolated groups from a number of different loci by comparing the gene trees. Different genes have different genealogies within a species due to recombination. It establishes gene flow delimiting species by identifying the unshared polymorphisms, and thus branches that are incompatible, with all genealogies at all loci.
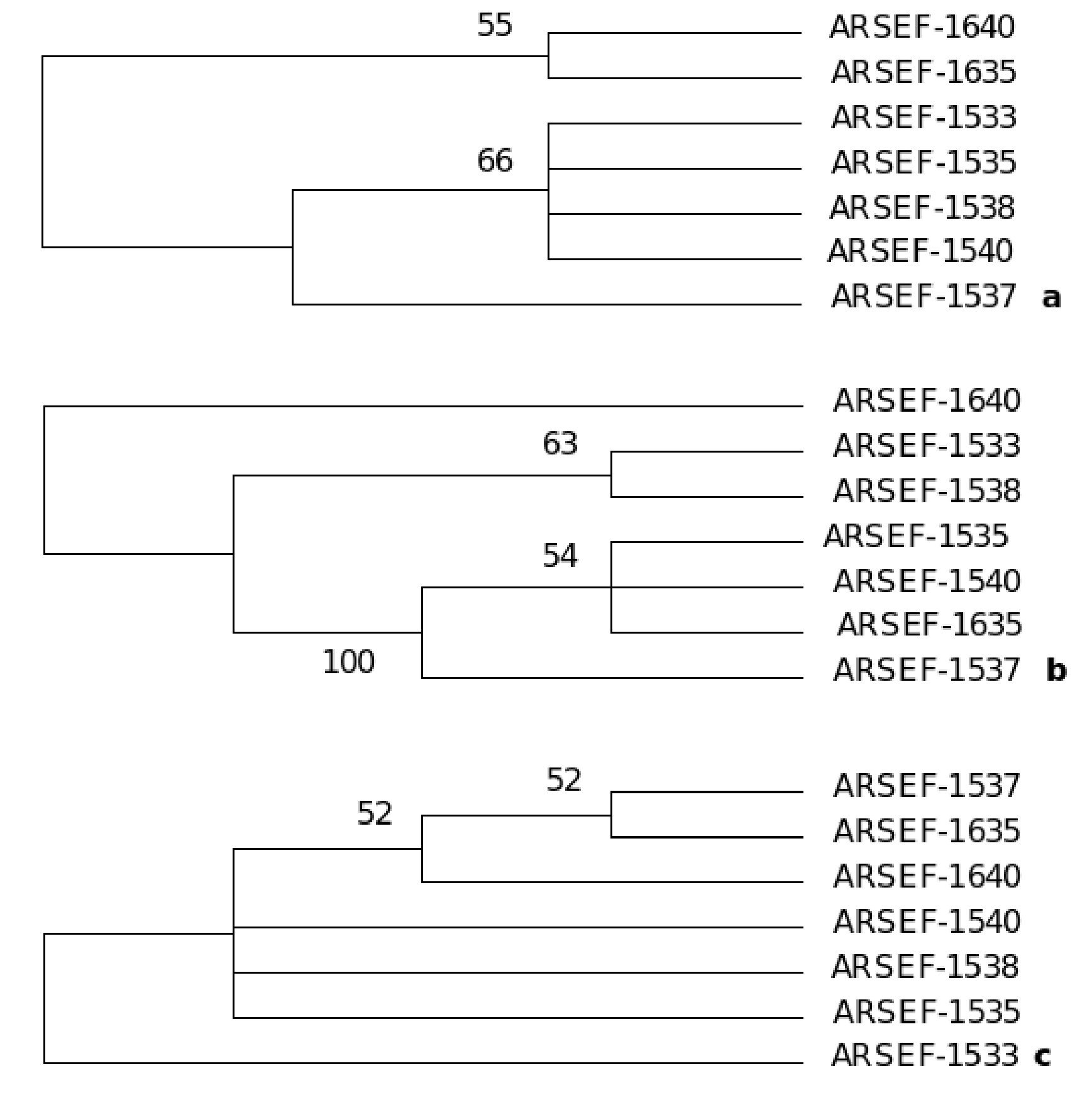
A consensus maximum parsimony tree was generated (PAUP ver. 4.0)31 from the partial sequences of the three genes: β-tubulin gene, large and small subunit of rRNA genes of mitochondria aligned by AlnExplorer (MEGA ver 3.1) derived from the isolates of an epizootic population of B. bassiana collected from Burgenland, Austria (Catalogue of the entomopathogenic fungi at ARSEF, Ithaca, New York). The details of the maximum parsimony tree generated from the aligned DNA sequences of each gene are as follows: β-tubulin gene: The tree was constructed from 296 characters, of which 285 were constant and 11 were variable, and of these 11 variable characters only one was parsimony informative (Fig. 12). The tree length (L) was 11, consistency index (CI) was 1.00, homoplasy index (HI) was 0.00 and retention index (RI) was 1.00.
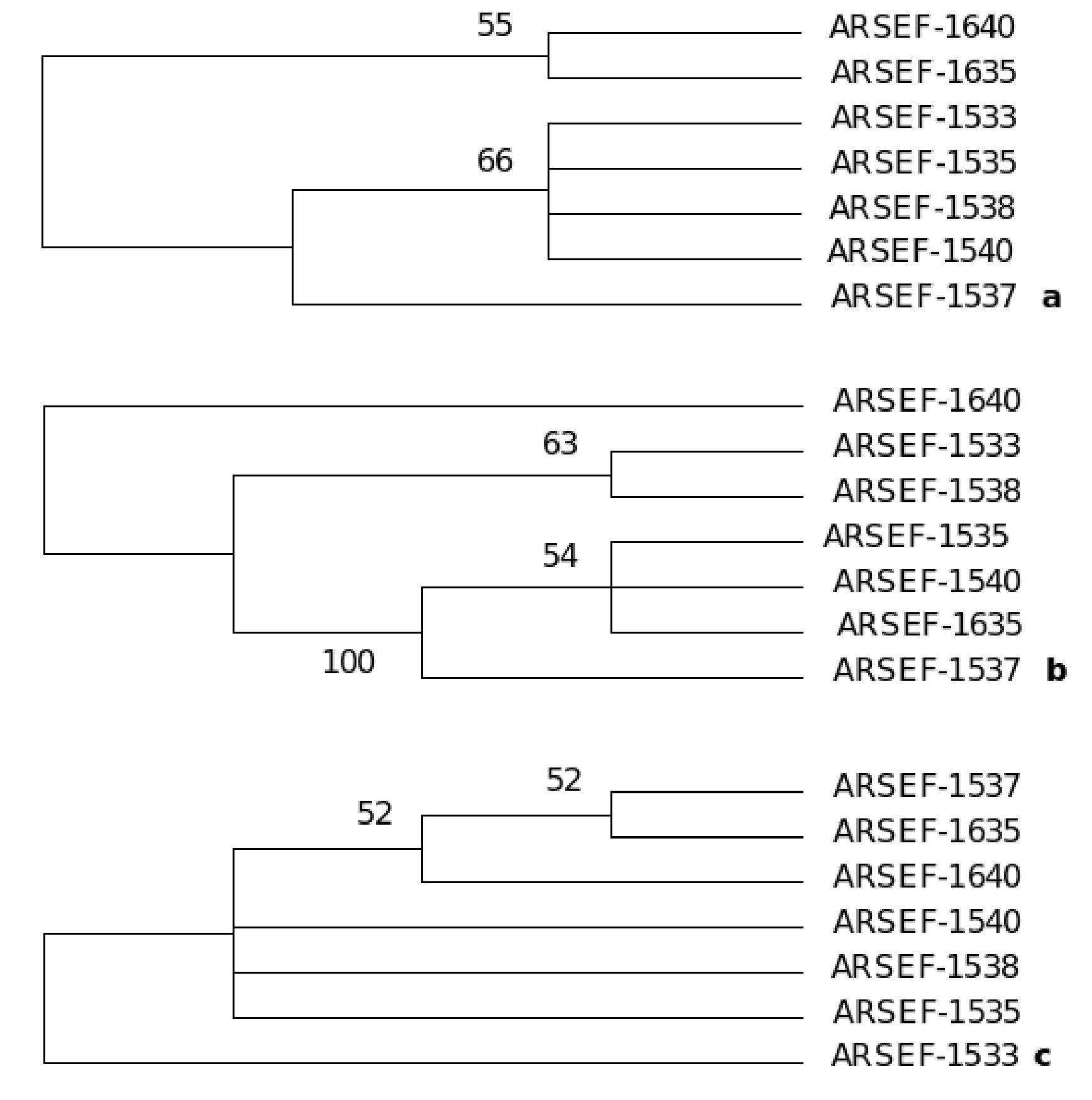
Figure 12. One consensus maximum parsimony tree generated from the sequences of (a) partial sequence of β-tubulin gene, (b) large subunit of mt rRNA gene and (c) small subunit of mt rRNA genes derived from the isolates of an epizootic B. bassiana population from Burgenland, Austria. Numbers below branches represent bootstrap values (%) based on 500 replicates. Only values >50% are shown. The tree topology of each species tree indicates the presence of cryptic speciation.
Gene for large subunit of rRNA of mitochondria: The tree was constructed from 183 characters, of which 105 were constant and 78 were variable and of these 37 were parsimony informative (Fig. 12). The tree length (L) was 109, consistency index (CI) was 0.89, homoplasy index (HI) was 0.10 and retention index (RI) was 0.80. Gene for small subunit of rRNA of mitochondria: The tree was constructed from 504 characters of which 498 were constant and six were variable and of these six variable characters one was parsimony informative (Fig. 9). The tree length (L) was 6, consistency index (CI) was 1.00, homoplasy index (HI) was 0.00 and retention index (RI) was 1.00.
The tree topology of each species tree indicates the presence of cryptic speciation. Incongruity of gene genealogies within a given group indicates gene flow and delimits a species. As the approach detects reproductive isolation, the resulting groups also fulfill the criteria of a biological species. Genealogical concordance phylogenetic species recognition (GCPSR) detects genetically isolated groups by comparing the gene trees from a number of different loci. Different genes have different genealogies within a species due to recombination. It establishes gene flow delimiting species by identifying the unshared polymorphisms, and thus branches that are incompatible with all genealogies at all loci, represent different species.
Discussion
The β-tubulin gene- and rRNA gene-based phylogenetic analysis affirmed the traditional taxonomic grouping of fungi, which was earlier substantiated by molecular phylogenetic studies17. The position of the entomopathogenic fungi B. bassiana, B. brongniarti, N. rileyi, M. anisopliae and Paecilomyces species in the family Clavicipitaceae of Hypocreales in Ascomycota announced in different phylogenetic analysis was found true.
The entomopathogenic fungi in the present sample have been shown earlier through molecular phylogenetic studies of samples with different fungal isolates to be closely related to another entomopathogenic mitosporic fungus - Lecanicillium lecanii and other species of Paecilomyces, Beauveria, Cordyceps and Nomuraea2,8,21,22,27,29,35. Thus, all the five important conidiogenous fungal genera with entomopathogenic habit - Beauveria, Nomuraea, Metarhizium, Paecilomyces and Lecanicillium -are closely related. Among these fungi, M. anisopliae diverged further from the rest of the entomopathogenic species and showed closer affiliation to sexual Clavicipitaceae both in the present study and in a previous analysis based on mitochondrial genes by Uribe and Khachatourians36 and rRNA gene of the small subunit22.
In the present analysis, the boundaries between species in the entomopathogenic fungal genera were nebulous. B. brongniartii and B. amorpha were found dispersed among the B. bassiana isolates though in a separate clade. A similar observation was made in the allozyme analysis of a very large sample of Beauveria species by St. Leger et al30 -some of the B. brongniartii isolates clustered separately while some grouped along with B. bassiana isolates. Phylogenetic analysis with the β-tubulin gene sequence not only iterated the complex nature of B. bassiana species27 but also revealed the great genetic distance between the members in the species, the distance being more than between morphologically distinguishable species. Boucias et al3 observed some N. rileyi isolates clustering with M. anisopliae. Such species overlapping was also observed in molecular phylogenetic studies with different members of this group21,22,31. These observations led to the conclusion that the asexual insect pathogenic fungi must have all diverged from a common claviciptalean ancestor. Loss of sex in these claviciptalean entomopathogenic fungi is believed to be a derived character2 having been attained simultaneously or subsequent to acquisition of entomopathogenic habit. Pathogenecity systems are believed to be flexible enough to continue to provide virulence against a broad range of hosts despite the absence of sexual recombination2. In fact, episodic events in host population dynamics may favour clonal populations because of the rapidity and low energy cost associated with asexuality; the population may indeed benefit from reduced out crossing through the maintenance of well-adapted pathotypes2. Muller's Ratchet effect - a population genetic mechanism that describes how asexual populations may undergo an unavoidable and progressive decline in fitness (due to absence of recombination mediated through sexual reproduction) eventually leading to extinction - was found to not apply to asexual entomopathogenic fungi2. Thus, the imperfect entomopathogenic fungal genera represent different mitotic forms of a perfect fungus of Clavicipitaceae.
Acknowledgements
NNRR and CUM are thankful to CSIR, New Delhi, for a research fellowship. We thank the DAAD-DST PPP-03 for facilitating this research collaboration.
* Corresponding author.
E-mail address: umadevikoduru@gmail.com (U.D. Koduru).
ARTICLE INFO
Article history:
Received June 18, 2007
Accepted September 9, 2008

