




Advances in the classification of the human pathogen Histoplasma capsulatum (H. capsulatum) (ascomycete) are sustained by the results of several genetic analyses that support the high diversity of this dimorphic fungus. The present mini-review highlights the great genetic plasticity of H. capsulatum. Important records with different molecular tools, mainly single- or multi-locus sequence analyses developed with this fungus, are discussed.
Recent phylogenetic data with a multi-locus sequence analysis using 5 polymorphic loci support a new clade and/or phylogenetic species of H. capsulatum for the Americas, which was associated with fungal isolates obtained from the migratory bat Tadarida brasiliensis.
This manuscript is part of the series of works presented at the “V International Workshop: Molecular genetic approaches to the study of human pathogenic fungi” (Oaxaca, Mexico, 2012).
Los resultados de diversos análisis genéticos que respaldan la alta diversidad de este hongo dimorfo confirman los progresos en la clasificación del patógeno humano Histoplasma capsulatum (H. capsulatum) (un ascomiceto). La presente revisión destaca la importante plasticidad genética de H. capsulatum. Se describen los datos importantes con los diferentes instrumentos moleculares, sobre todo, los análisis de las secuencias individuales o multi-loci establecidos con este hongo.
Datos filogenéticos recientes con un análisis multi-loci de secuencias utilizando 5 loci polimorfos respaldan un nuevo clado y/o especie filogenética de H. capsulatum del continente americano, asociado a aislamientos fúngicos obtenidos del murciélago migratorio Tadarida brasiliensis.
Este artículo forma parte de una serie de estudios presentados en el «V International Workshop: Molecular genetic approaches to the study of human pathogenic fungi» (Oaxaca, México, 2012).
The scientific history of the pathogenic fungus Histoplasma capsulatum began with the histopathological findings, published by Samuel Taylor Darling in 1906, in tissues of a patient from Martinique, who was working on the construction of the Panama Channel. Darling observed intracellular parasites with 1–6μm in diameter surrounded by a translucent halo.5 Due to its resemblance to Leishmania, it was described as a protozoan, and was named “H. capsulatum” because of the similarity of its halo with a capsule. Later, in 1912, Henrique da Rocha Lima inferred the mycotic nature of this pathogen, characterizing it as a yeast.6 The fungus is a saprobe-geophilic organism that utilizes two nomenclatures depending on its sexual state: H. capsulatum (anamorph or asexual state) and Ajellomyces capsulatus (teleomorph or sexual state); both constitute the same holomorph organism, which is the causative agent of histoplasmosis, a systemic mycosis with primary respiratory compromise.
Before the description of the sexual state of H. capsulatum by Kown-Chung,18,19 this pathogen was classified into the Division Deuteromycota, Order Moniliales, and Family Moniliaceae, based on the morphological criteria proposed in 1899 by Saccardo.24 Currently, H. capsulatum is classified into the Kingdom Fungi, Subkingdom Dikarya, Phylum Ascomycota, Class Eurotiomycetes, Order Onygenales, and Family Onygenaceae and/or Ajellomycetaceae,10–12 according to the analyses of six loci: 18S, 5.8S and 28S of the rRNA genes; EF1α (Elongation factor-1α); RPB1and RPB2 (RNA polymerase II subunits 1 and 2).
In the environment, this organism grows preferentially in bat and bird guano that contains high concentrations of nitrogen and phosphorus in addition to other micronutrients. These conditions, together with optimal air and soil temperatures (18–28°C), humidity (>60%), and darkness (fosters sporulation), characterize the ideal ecological niche of H. capsulatum, which favors the development of its multicellular infective mycelial phase (M-phase).27,29,34–36
Classification of Histoplasma capsulatumThe biological species H. capsulatum comprised three taxonomic varieties: H. capsulatum var. capsulatum Darling, 1906; H. capsulatum var. duboisii (Vanbreuseghem, 1957) – Ciferri, 1960; and H. capsulatum var. farciminosum (Rivolta, 1873) – Weeks, Padhye, et Ajello, 1985. These varieties were identified by their micromorphologies, geographic distribution, host-association, and clinical forms of the disease. Currently, with the advent of molecular techniques to classify fungal species, these taxonomic varieties have been included in a molecular taxonomy based on the phylogenetic species concept.15
Taylor et al.33 indicated that the concepts and/or criteria of species recognition often used in mycology are the biological and morphological ones, and most of the described species have been identified with phenotypic characters. However, some pathogenic fungi show few informative characters thereby leading to skewed, controversial, and erroneous classifications.8,9 To overcome these inconveniences, most authors have promoted genetic and molecular statements for genotypic and phylogenetic classifications of pathogenic fungi.
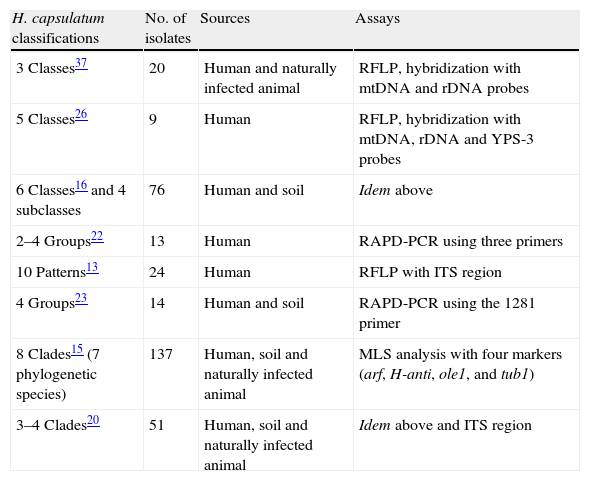
Genotyping of Histoplasma capsulatumH. capsulatum isolates have been grouped based on their genotype patterns using different molecular assays (Table 1). First, Vincent et al.37 grouped clinical strains in three classes according to their Restriction Fragment Length Polymorphism (RFLP) profiles and hybridization with mtDNA and rDNA probes. Class 1 included only the Downs strain of H. capsulatum var. capsulatum from North America; Class 2 was formed by 14 H. capsulatum var. capsulatum strains from North America and two H. capsulatum var. duboisii strains from Africa; finally, Class 3 grouped four H. capsulatum var. capsulatum strains from Central America and two H. capsulatum var. capsulatum strains from South America. Spitzer et al.,25 using the same methodology, proposed a new Class 4; one year later, a Class 5 based on hybridization with a probe of the YPS-3 gene was recorded.26
Relevant molecular classifications of Histoplasma capsulatum isolates.
| H. capsulatum classifications | No. of isolates | Sources | Assays |
| 3 Classes37 | 20 | Human and naturally infected animal | RFLP, hybridization with mtDNA and rDNA probes |
| 5 Classes26 | 9 | Human | RFLP, hybridization with mtDNA, rDNA and YPS-3 probes |
| 6 Classes16 and 4 subclasses | 76 | Human and soil | Idem above |
| 2–4 Groups22 | 13 | Human | RAPD-PCR using three primers |
| 10 Patterns13 | 24 | Human | RFLP with ITS region |
| 4 Groups23 | 14 | Human and soil | RAPD-PCR using the 1281 primer |
| 8 Clades15 (7 phylogenetic species) | 137 | Human, soil and naturally infected animal | MLS analysis with four markers (arf, H-anti, ole1, and tub1) |
| 3–4 Clades20 | 51 | Human, soil and naturally infected animal | Idem above and ITS region |
Keath et al.,16 in accordance with Spitzer et al.,26 increased the number of strains studied and expanded the fungus genotyping to six classes, including four subclasses within Class 5. It is noteworthy that, from the initial proposal of Vincent et al.37 until the classification of Keath et al.,16 most of the studied strains came from restricted geographic areas of North America, and very few strains were isolated in Central (Panama) and South America (Colombia).
Later, Poonwan et al.,22 analyzed 13 clinical isolates of H. capsulatum from Thailand with Random Amplification of Polymorphic DNA (RAPD-PCR), using three oligonucleotides separately, and found that isolates from Thailand formed two to four homogeneous groups that were clearly separated from the G-217B reference strain from North America.
Jiang et al.13 genotyped 24 fungal isolates from the United States of America through the nucleotide sequence analysis of the ITS1-5.8S-ITS2 region of the rDNA, and found 10 different H. capsulatum sequence patterns. As a result, they suggested that this method could be useful for reorganizing isolates from other classifications.
Based on a RAPD-PCR assay with the single random primer 1281, Reyes-Montes et al.23 distinguished four groups (I–IV) and two subgroups (Ia and Ib) of H. capsulatum from different origins in Latin America (Mexico, Guatemala, Panama, and Colombia), which were isolated from clinical and environmental sources. In this study, the reference strain G-186B from Panama (Class 3, according to Vincent et al.37) formed a single group. The latter authors suggested that RAPD-PCR profiles with suitable random primers could be used for classifying fungal isolates in accordance with their source and geographic distribution. The RAPD-PCR method with four independent random primers was used by Muniz et al.21 to analyze 48 H. capsulatum samples from Rio de Janeiro State (Brazil) isolated from different sources (soil, animal, and human clinical samples), which were grouped according to the genetic polymorphism generated for each primer. The RAPD-PCR profiles were able to separate the Brazilian isolates from the North American (United States of America) isolates in accordance with their percentage of similarity. Afterwards, Zancopé-Oliveira et al.,38 using a similar procedure, analyzed 22 Brazilian H. capsulatum isolates, mostly from human clinical samples, and identified three clusters: cluster I, with isolates from the Brazilian north-eastern region; a major cluster II, with isolates from the Brazilian south-eastern and south regions; and cluster III (48% similarity), with isolates from Goias State in the central region of Brazil.
Additional molecular studies by Carter et al.,2–4 Kasuga et al.,14 and Taylor et al.31 have referred differences in the population structures of H. capsulatum (clonal and recombinant). The analyses of H. capsulatum genetic populations, mainly with the (GA)n, (GT)n and GT(A)n multiallelic markers (microsatellites), enabled the possibility of distinguishing H. capsulatum isolates from the United States of America and Colombia, suggesting the separation of these fungal populations in distinct phylogenetic species.2,4 Recently, Taylor et al.,32 based on the sequences of a 240-nt fragment of the (GA)n microsatellite and its flanking regions, found two major clusters, I and II, according to the genetic diversity of H. capsulatum isolated from nine distinct bat species captured in different geographic regions from Mexico and Brazil, emphasizing a sub-cluster grouped in cluster I that carried a unique haplotype of H. capsulatum samples that were isolated from the migratory bat Tadarida brasiliensis.
Today, advances in fungal classification are supported by robust analyses of the sequences of gene fragments that provide important genetic informative sites to separate clusters of isolates. The statement of a large number of these sites associated with several loci would establish a clear relatedness among fungi and would contribute to a better understanding of the intra- and inter-specific diversity of fungal species. In the last ten years, Multi-Locus Sequence Typing (MLST) has been used to group or classify pathogenic fungi.30 MLST or Multi-Locus Sequence (MLS) analyses utilize sequences of several genes with sufficient polymorphism for differentiating isolates of the same fungal species. It is an excellent molecular tool to characterize genetic diversity in different microorganisms.
Phylogenetic classification of Histoplasma capsulatum: Multi-Locus Sequence analysesIn some circumstances, the molecular markers have been useful for identifying species that remained cryptic under morphological and/or biological criteria. Currently, the identification of these species are revealed by phylogenetic, genealogic, or gene concordance species concepts, which group the organisms according to the changes in their nucleic acids.8,9,33 Taking into consideration the aforementioned concepts, H. capsulatum must be considered a cryptic phylogenetic species complex.33
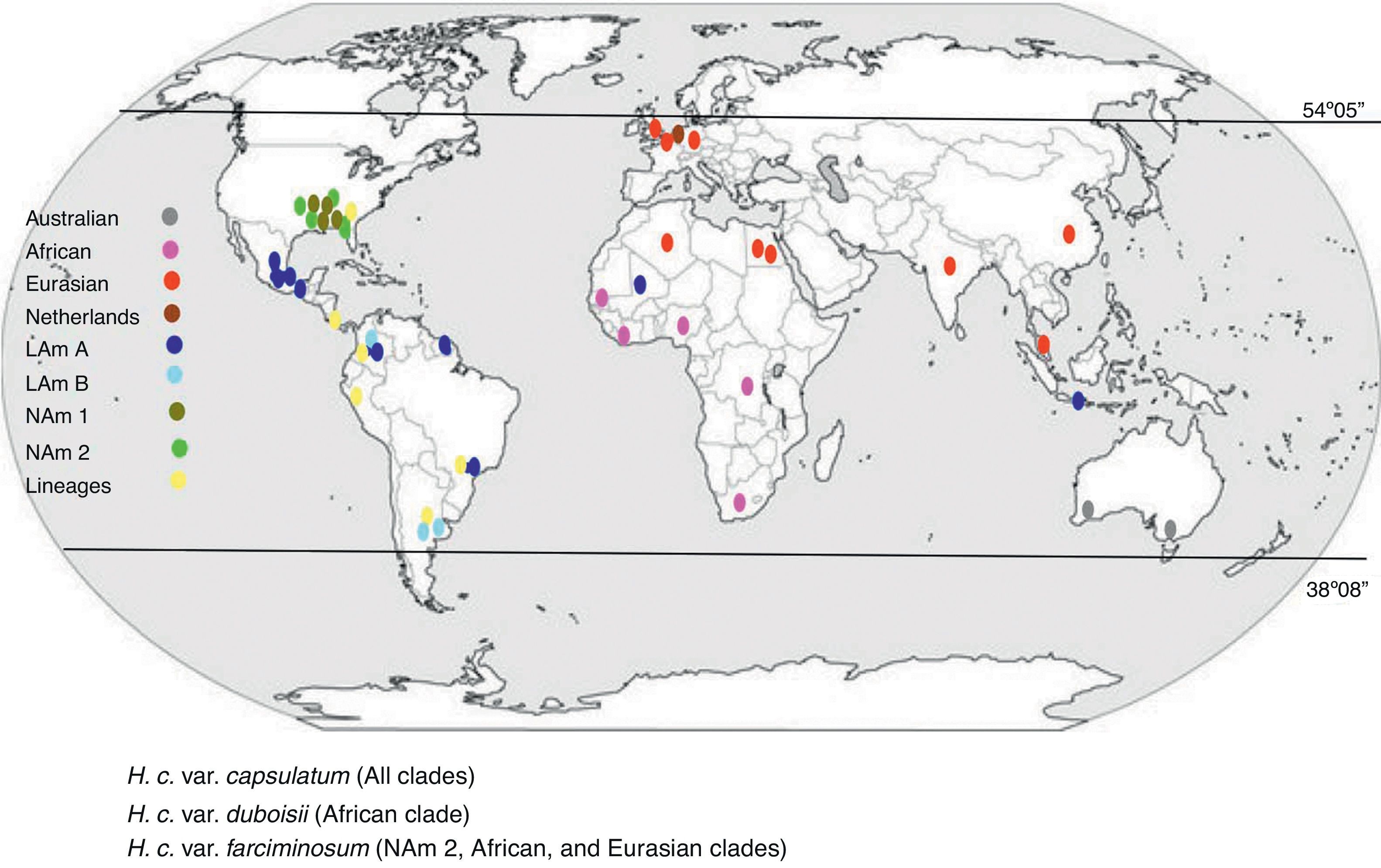
Two pioneer MLS studies, which contributed to the phylogenetic classification of H. capsulatum, were reported by Kasuga et al.14,15 (Table 1). In the last report, using 137 isolates from 25 countries, Kasuga et al.15 proposed a phylogeographic classification of H. capsulatum based on the partial sequences of four protein-coding genes (arf, H-anti, ole1, and tub1), which include eight genetic populations (clades) named as: North America class 1 (NAm 1), North America class 2 (NAm 2), Latin America group A (LAm A), Latin America group B (LAm B), Australian, Netherlands, Eurasian, and African. With the exception of the Eurasian clade, seven clades were considered to be phylogenetic species. Consistent with the Kasuga et al.15 data, Fig. 1 was constructed to represent the H. capsulatum clades and lineages around the world.

Geographic distribution of the H. capsulatum clades and lineages based on their actual phylogeographic classification. Data were obtained from Kasuga et al.,15 who proposed this classification based on the partial sequences of four protein-coding genes, using 137 fungal isolates from 25 countries; most were of the taxonomic variety H. capsulatum var. capsulatum. Some individuals of the taxonomic varieties H. capsulatum var. duboisii and H. capsulatum var. farciminosum were also included in different clades.
Following the Kasuga et al.14,15 contributions, several authors have used the same gene fragments to advance in the phylogenetic study of H. capsulatum. Taylor et al.,28 conducted an MLS analysis with 14 isolates of the fungus obtained from naturally infected bats captured in Mexico with different migratory habits (Artibeus hirsutus-non-migratory and Leptonycteris nivalis, Leptonycteris curasoae, T. brasiliensis-migratory), which suggested the existence of a new clade of H. capsulatum related to isolates from the infected T. brasiliensis bats. Later, Muniz et al.,20 using the same aforementioned protein-coding genes, analyzed 51 isolates of H. capsulatum from different sources and Brazilian geographic areas, and revealed three to four clades according to the phylogenetic trees generated; furthermore, the authors suggested different population structures in the isolates from Brazil. Finally, Balajee et al.1 conducted an MLS analysis with arf, H-anti, and tub1 gene fragments, and the ITS region. These authors extracted the DNA of H. capsulatum from paraffin-embedded tissue samples from cats diagnosed with histoplasmosis, living in three cities localized in non-endemic areas of the disease in the United States of America. This study included, as reference, 82 sequences of the fungal strains previously reported by Kasuga et al.15 representing their eight described clades, and the authors suggested that their results demonstrated a new phylogenetic clade associated with all H. capsulatum strains causing cat infections.
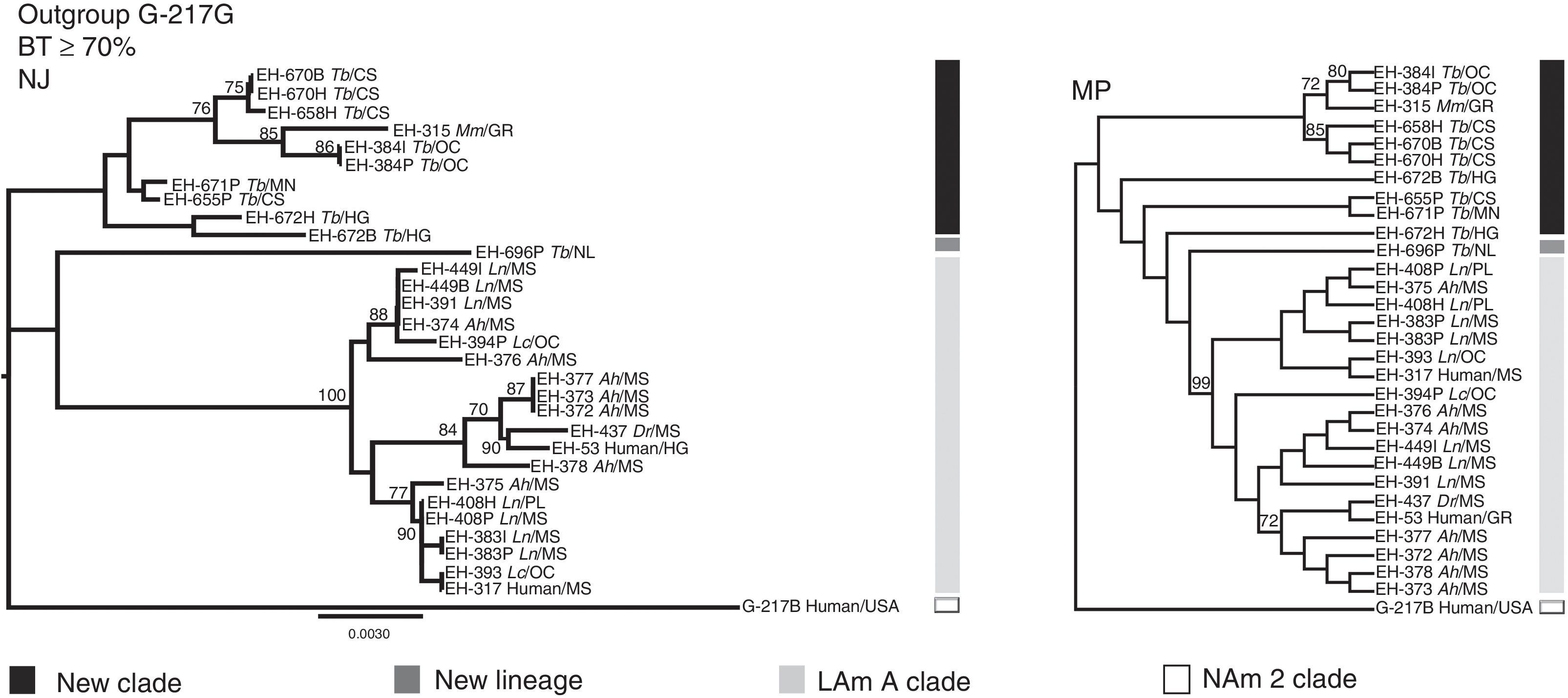
Based on unpublished results from our laboratory, with 28 H. capsulatum isolates obtained from wild bats captured in Mexico and two reference strains from Mexican histoplasmosis patients, it was possible to generate more robust data to classify these fungal samples by using an MLS analysis with various informative sites of highly polymorphic loci [arf, H-anti, ole1, tub1, and the (GA)n microsatellite]. Similarities and genetic distances among their sequences demonstrated that fungal isolates recovered from cave-dwelling bats and the two clinical reference strains from the central states of Mexico (Morelos, Puebla, and Hidalgo) were grouped independently from most H. capsulatum isolated from the migratory bat T. brasiliensis (captured in the Mexican states of Chiapas, Hidalgo, and Michoacán) and from one isolate from a Mormoops megalophylla bat captured in Guerrero (Fig. 2). This finding matches previous data published by Taylor et al.,28,32 who had proposed that fungal isolates from T. brasiliensis form a new clade because these isolates did not group with any isolate from the Kasuga et al.15 clades. In addition, a single H. capsulatum isolate from a T. brasiliensis captured in the Northwest of Mexico (Nuevo León State) forms a new lineage. Thus, the topology of the two concatenated trees that were generated by an MLS analysis using the five aforementioned polymorphic loci were able to discriminate the high genetic diversity of this fungus, thereby contributing to the knowledge of the pathogen (Fig. 2).
n microsatellite. Phylogenetic analyses were conducted as follows: (1) the neighbor-joining (NJ) method using the Kimura two-parameter model17; and (2) the maximum parsimony (MP) method using the close-neighbor-interchange algorithm.7 Data of parsimony were: MPT=99; LT=188; CI=0.584615; RI=0.881569. Trees were generated with 1000 replicates. Fourteen of the Mexican H. capsulatum isolates were previously classified by Kasuga et al.15 as belonging to the LAm A clade. Abbreviations: Tb (T. brasiliensis), Ah (A. hirsutus), Ln (L. nivalis), Lc (L. curasoae), Mm (M. megalophylla), Dr (Desmodus rotundus), CS (Chiapas), GR (Guerrero), HG (Hidalgo), Mn (Michoacán), MS (Morelos), NL (Nuevo León), OC (Oaxaca), PL (Puebla), and USA (United States of America).")
Concatenated phylogenetic trees of Mexican H. capsulatum isolates using an MLS analysis. Trees were constructed with MEGA5 software considering the sequences, from 30 H. capsulatum samples, of the polymorphic loci: arf, H-anti, ole1, tub1, and the (GA)n microsatellite. Phylogenetic analyses were conducted as follows: (1) the neighbor-joining (NJ) method using the Kimura two-parameter model17; and (2) the maximum parsimony (MP) method using the close-neighbor-interchange algorithm.7 Data of parsimony were: MPT=99; LT=188; CI=0.584615; RI=0.881569. Trees were generated with 1000 replicates. Fourteen of the Mexican H. capsulatum isolates were previously classified by Kasuga et al.15 as belonging to the LAm A clade. Abbreviations: Tb (T. brasiliensis), Ah (A. hirsutus), Ln (L. nivalis), Lc (L. curasoae), Mm (M. megalophylla), Dr (Desmodus rotundus), CS (Chiapas), GR (Guerrero), HG (Hidalgo), Mn (Michoacán), MS (Morelos), NL (Nuevo León), OC (Oaxaca), PL (Puebla), and USA (United States of America).
The authors declare that they have no conflict of interests.
This paper constitutes partial fulfillment of the requirements of the Graduate Program in Biological Sciences of the UNAM. TVG thanks the scholarship No. 324232 provided by the National Council of Science and Technology (CONACyT).

