



Disorders of sex development (DSD), which has the possibility of the risk of life-threatening endocrinologic emergencies of the newborn, require a careful multidisciplinary approach.
ObjectivesThe aim of our study is to consolidate the proposed classification, evaluation and management of DSD.
Materials and methodsThe literatures related with DSD were reviewed to find the best approach for this disease.
ResultsThe detailed history, systemic physical examination of the patient, particular laboratory and imagine evaluations are needed for the urgent treatment of life-threatening abnormalities and the gender assignment.
ConclusionThe gender should be assigned depending on the definitive diagnosis, fertility potential, genital appearance, surgical options, and the parents’ opinion.
Los trastornos del desarrollo sexual (TDS), que tienen la posibilidad de poner en riesgo vital las emergencias endocrinológicas del recién nacido, precisan un cuidadoso enfoque multidisciplinar.
ObjetivosEl objetivo de nuestro estudio es consolidar la propuesta de clasificación, evaluación y tratamiento de los TDS.
Materiales y métodosSe revisaron las literaturas relativas a TDS, a fin de hallar el mejor enfoque para esta enfermedad.
ResultadosSe precisan la historia clínica detallada, la exploración física sistémica del paciente, el laboratorio concreto y las evaluaciones de imágenes para tratar urgentemente las anormalidades con riesgo vital y la asignación de género.
ConclusiónDeberá asignarse el género dependiendo del diagnóstico definitivo, el potencial de fertilidad, el aspecto de los genitales, las opciones quirúrgicas y la opinión de los padres.
Sex development disorder occurs in 1–2 of every 10,000 newborns.1 Ambiguous genitalia are a serious concern for the family and the first physician examining the child. This is due to not only ambiguous genitalia, but also the risk of life-threatening endocrinologic emergencies of the newborn. Therefore, ambiguous genitalia require a careful multidisciplinary approach. Evaluation and management should be performed in a center with a multidisciplinary team, comprised of pediatric specialists in surgery, typically in urology, endocrinology, neonatology, nursing, psychology, genetics, and medical ethics.1–5
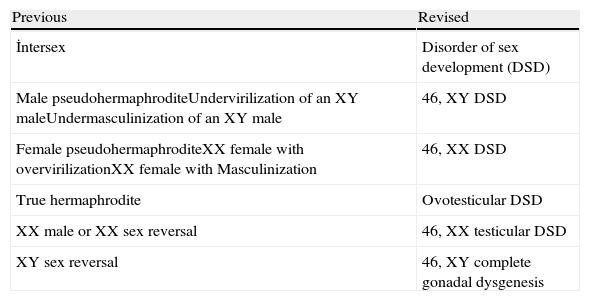
ObjectivesDesignations such as hermaphroditism or pseudohermaphroditism, which are previous terminologies, are confusing to patients and their relatives, and also constitute a humiliating situation. Therefore, the term “disorders of sex development” (DSD) is proposed to indicate congenital conditions with atypical development of chromosomal, gonadal, or anatomic sex. This terminology should be well-grounded, open to new information, and explain the genetic etiology and phenotypic changes, comprehensible by the physician, patient, and the parents.
This study was designed to consolidate the proposed new classification, and to facilitate the evaluation and the management of DSD.
Materials and methodsThe literatures related with DSD were reviewed to find the best approach for this disease. An extensive research was in the Pubmed. The MeSH terms, such as “hermaphroditism”, “pseudohermaphroditism”, “disorders of sex development”, “congenital adrenal hyperplasia” and “androgen insensitivity syndrome”, were used in research of the articles without the limits of publication date.
ResultsIn 2006, under the sponsorship of the European Society for Pediatric Endocrinology (ESPE) and the Lawson Wilkins Pediatric Endocrine Society (LWPES), with the participation of 50 experts in Chicago, the DSD results were evaluated and published as a consensus.6 Afterwards, the DSD consensus was reviewed with the current practical applications of 60 centers from 23 European countries and the following topics were emphasized: the necessity of the evaluation of patients by skilled professionals, avoidance of gender assignment in the newborn without the assessment of an expert, the need for a multidisciplinary team, psychosocial support for the patients, surgical procedures performed by the surgeons specialized in this field, assessment of the functional and the cosmetic surgical procedures all together, and more accurate information for the parents and the patients, which will lead to a definitive final decision. In order to define the disease more accurately and clearly, today, new nomenclature and classification are used (Tables 1 and 2).
Old and new nomenclatures.
| Previous | Revised |
| İntersex | Disorder of sex development (DSD) |
| Male pseudohermaphroditeUndervirilization of an XY maleUndermasculinization of an XY male | 46, XY DSD |
| Female pseudohermaphroditeXX female with overvirilizationXX female with Masculinization | 46, XX DSD |
| True hermaphrodite | Ovotesticular DSD |
| XX male or XX sex reversal | 46, XX testicular DSD |
| XY sex reversal | 46, XY complete gonadal dysgenesis |
Proposed classification of DSD.
| Sex chromosome DSD(variable) | 46, XY DSD(under-virilized genetic male) | 46, XX DSD(over-virilized genetic female) |
| A: 47,XXY (Klinefelter Syndrome and variants)B: 45,X (Turner Syndrome and variants)C: 45, X/46, XY (mixed gonadal dysgenesis)D: 46, XX/46, XY (chimeric, ovotesticular DSD) | A: Disorders of testicular development1. Complete or partial gonadal dysgenesis (e.g. SRY, SOX9, SFI, WT1, DHH, etc.)2. Testis regressionOvotesticular DSDB: Disorders in androgen synthesis/actionAndrogen biosynthesis defect(e.g. 17-hydoxysteroid dehydrogenasedeficiency, 5a reductase deficiency, StAR mutations)Defect in androgen action(e.g. CAIS, PAIS, drugs)LH receptor defects (e.g. Leydig cell Aplasia, hypoplasia)Disorders of AMH and receptor(Persistent Mullerian Duct Syndrome)C: Other (e.g. cloacal extrophy, severe hypospadias) | A: Disorders of ovarian development1. Testicular DSD (e.g. SRYş, dup SOX9, RSP01)2. Ovotesticular DSDGonadal dysgenesisB: Androgen excessFetal (e.g. 21 hydroxylase deficiency, 11 hydroxylase deficiency, glucocorticoid receptor mutations)Fetoplacental (aromatase deficiency, oxidoreductase deficiency-POR)Maternal (luteoma, Androgenic drugs, etc.)C: Other (e.g. cloacal extrophy, vaginal atresia, MURCS, labial adhesions, other syndromes) |
The male and female embryos are phenotypically identical until the sixth gestational week in the normal conditions. The gonads are bipotential at first, forming as a mesodermal thickening in the urogenital ridge. There are both Müllerian and Wolffian ducts. In the presence of 46, XY karyotype bipotential gonad differentiates into testis. If the karyotype is 46, XX the differentiation progresses in the favor of ovary.7 There are number of genetic factors affecting this sex-determining process.
In the presence of a Y chromosome, the Sertoli cells express the sex-determining region Y gene (SRY) that encodes the testis-determining factor. A number of downstream gene expressions are triggered by SRY and as a result, cell types further differentiate into the testis.8 The Wolffian ducts develop into male internal genitalia (epididymis, vas deferens, and vesicula seminalis) by the effect of testosterone produced by Leydig cells. Dihydrotestosterone (DHT) is formed from testosterone by the enzyme 5α-reductase. DHT is necessary for the development of external male genitalia.9 Sertoli cells also produce anti-Müllerian hormone (AMH) which leads to the involution of the Müllerian structures.10
In the case of ovarian differentiation, it begins a week later and requires a number of specific factors. In the XX embryos, AMH and testosterone are absent, and thus the Müllerian ducts develop into the internal female genitalia (the uterus, Fallopian tubes, and the upper one-third of the vagina). Due to the absence of testosterone, the Wolffian ducts begin to regression.
Psychosexual development, androgen exposure, and the sex chromosome genes together with the central nervous system, affect social circumstances and family dynamics. Sexual identity is to define oneself as male or female. Sex role defines gender-specific behavioral and psychological characteristics. Sexuality is dimorphic as toy selection and physical aggression. Sexual tendency determines the orientation of erotic interest (homosexual, heterosexual, bisexual), and includes behavior, fantasy or attractiveness.
Genitalia, size of phallus, scrotum, labioscrotal fusion, palpation of gonads, genital or areolar pigmentation, and meatal and vaginal apertures should be carefully observed in addition to a complete physical examination.
In the evaluation of DSD in female newborns, marked ambiguous genitalia, posterior labial fusion, inguinal or labial mass and inguinal hernia with a gonad should be considered. In male newborns, micropenis, non-palpable testis, isolated perineal hypospadias and marked hypospadias with undescended testis should cause suspicion. Additionally, positive family history and discordance between genitalia and karyotype are important parameters supporting the diagnosis.
Delayed presentations can appear with a various manifestations: the inability to adapt grown sex, sexual dysfunction due to narrow vagina or small penis, precocious puberty [XY-congenital adrenal hyperplasia (CAH)], behavioral disorders (5α-reductase deficiency), infertility, recurrent hematuria, neoplastic gonad, delayed puberty, primary amenorrhea [complete androgen insensitivity syndrome (CAIS)], and breast development in males and inguinal hernias in females.
There are some important points to be aware of in the early and delayed evaluation of patients with DSD. In CAH, salt and electrolyte imbalance (adrenal crisis) is a urological emergency that should be diagnosed and treated immediately.11 Pathology that leads to DSD should be detected, fertility should be maintained and sexual identification and assignment should be done in a satisfactory way. The concern of the family should be decreased while guiding the patient to be a sexually and socially functional individual, and on the other hand, the potential for the development of a malignancy should be avoided.
Evaluation of DSDWhile evaluating patients with DSD, a detailed history of pregnancy should be obtained as well as the medications taken during pregnancy (exposure to androgens). The previous diseases should also be thoroughly questioned. The blood relationship of the parents, neonatal deaths, DSD disorders and genital anomalies in the previous births should carefully be investigated. The history of primary amenorrhea and infertility in other family members, newborn growth failure, persistent vomiting, and diarrhea should be investigated to rule out a maternal androgen-producing tumors.
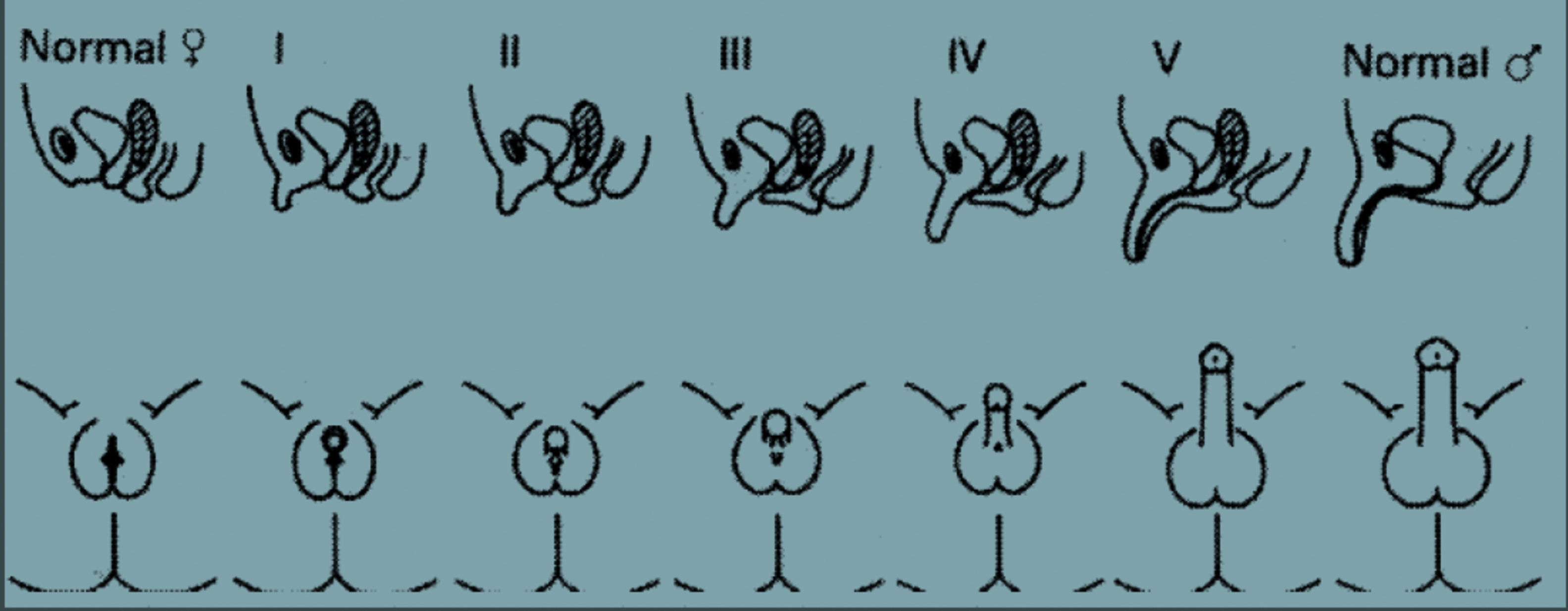
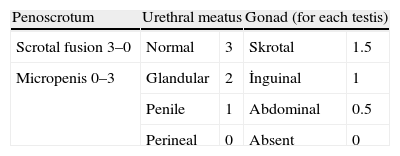
Systemic physical examinations must be carried out. Arterial blood pressure monitoring, the presence of pigmentation in the genital and areolar area, the presence of hypospadias or urogenital sinus, the phallus size (in full-term newborns, prolonged penile length should be measured at least 2cm), and labioscrotal folds should be inspected carefully for symmetry and fullness. When there is asymmetry in these folds, usually a gonad is palpated on the side of virilization, which is frequently associated with inguinal hernias. The Prader staging system and external masculinization score are used for the evaluation of the external genital organs (Fig. 1 and Table 3).
. “Der genitalbefund beim Pseudohermaproditismus femininus des kongenitalen adrenogenitalen Syndroms. Morphologie, Hausfigkeit, Entwicklung und Vererbung der verschiedenen enitalformen. Helv. Pediatr. Acta. 9: 231-248.). Stage 0: normal female anatomy, Stage 1: enlarged phallus, Stage 2: extended phallus with separated urethral and vaginal openings, Stage 3: extended phallus with unilateral urogenital sinus opening, Stage 4: extended phallus with hypospadias, Stage 5: normal male anatomy.")
Staging system of Prader (Prader Von, A. (1954). “Der genitalbefund beim Pseudohermaproditismus femininus des kongenitalen adrenogenitalen Syndroms. Morphologie, Hausfigkeit, Entwicklung und Vererbung der verschiedenen enitalformen. Helv. Pediatr. Acta. 9: 231-248.). Stage 0: normal female anatomy, Stage 1: enlarged phallus, Stage 2: extended phallus with separated urethral and vaginal openings, Stage 3: extended phallus with unilateral urogenital sinus opening, Stage 4: extended phallus with hypospadias, Stage 5: normal male anatomy.
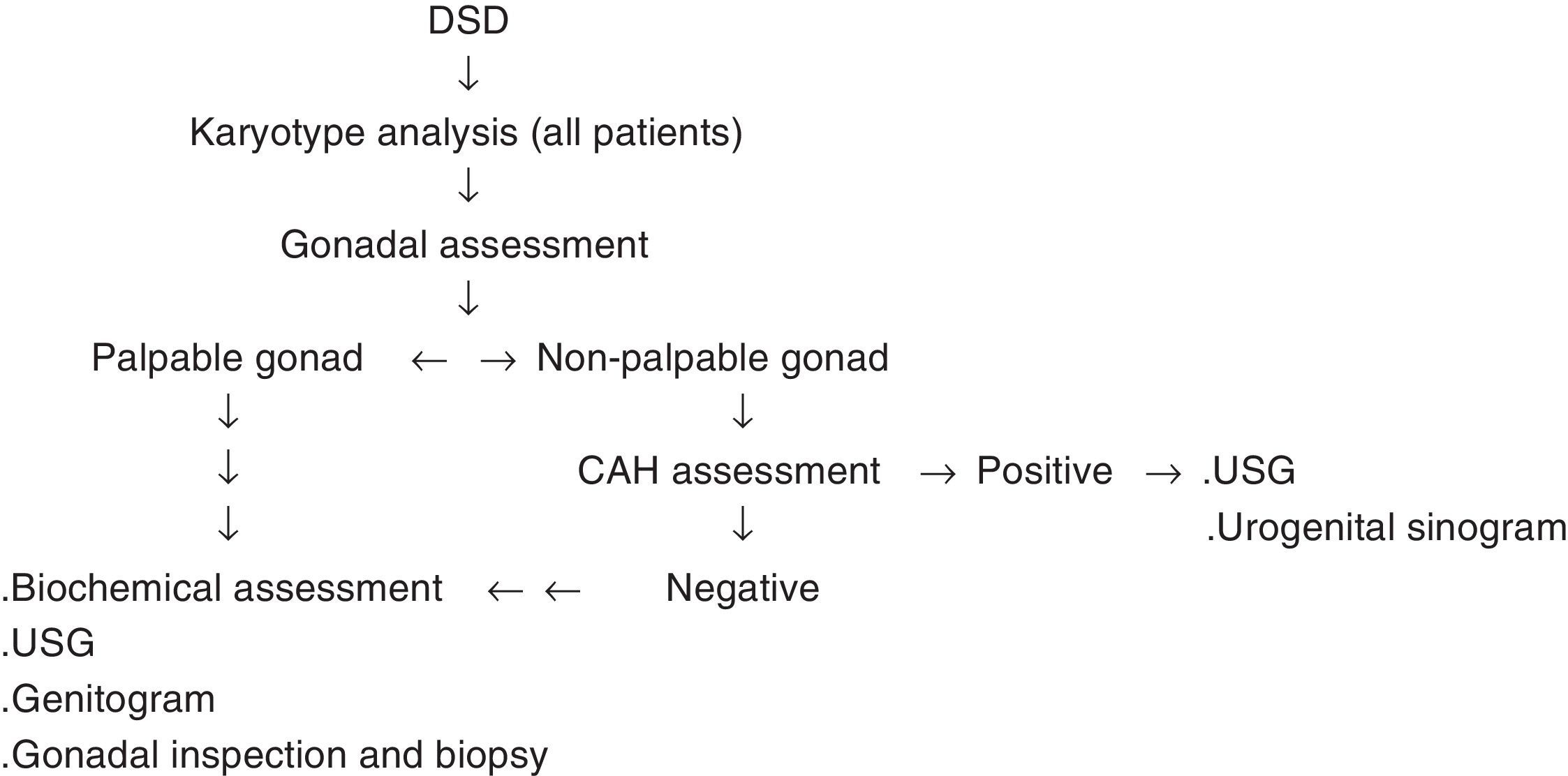
The clues obtained from history and physical examinations are combined with the laboratory and imaging methods in order to reach the definite diagnosis. The algorithm prepared by the American Academy of Pediatrics can also be used as a guide (Fig. 2).

All laboratory and imaging methods that can be used are as follows: cheek swab; blood analyses for 17-hydroxyprogesterone, testosterone, AMH, androstenedione, electrolytes, LH, FSH, cortisol, and ACTH; urine analyses for adrenal steroids; chromosome analysis, such as FISH analysis for the X and Y chromosomes, full karyotype analysis of peripheral venous blood, mosaicism (blood, genital skin, gonads); abdomino-pelvic ultrasonography; genitogram; HCG and ACTH stimulation test; androgen binding studies; and endoscopy.12
During the assessment, primarily the X and Y karyotype analyses should be conducted. Biochemical tests of blood and urine should be performed. In particular, 17-hydroxyprogesterone, testosterone, gonadotrophins, AMH, electrolytes, cortisol, ACTH, the 21-hydroxylase enzyme deficiency, which is the most common cause of CAH (90%), and 11-B-hydroxylase levels, the other rare cause of CAH (5%), should be assessed.13 Ultrasound, MRI, urogenital sinogram, panendoscopy, laparoscopy, biopsies of genital skin and gonadal androgen binding studies should be done according to the needs of the cases.
Testosterone response to the HCG test (baseline T, DHT, DHEA and androstenedione measurement followed by the administration of 3X500-1500IU HCG (IM) and after 24h the re-measurement of T, DHT, DHEA and androstenedione levels), a synacten test and androgen binding measurements are the other ancillary tests that can be used. Chromosome scans can be performed with specific mutational gene analyses (SRD5A2, AR, AMH, AMHR, CYP21, HSD17B3), DNA analysis for rare cases (DAKS1, SOKS9, WT1), and genomic DNA mutation analyses (DGGE, SSCP).
Specific molecular diagnosis can be made in 20% of the patients with DSD. In only half of the children with 46, XY DSD, a definitive diagnosis can be established, while most of the patients with 46, XX virilized CAH can be diagnosed through increased 17 hydroxyprogesterone, and androstenedione levels.1,14,15 The algorithm mentioned earlier can be utilized to establish the diagnosis (Fig. 2).
DiscussionGender determinationThe final point in DSD is the gender assignment. The gender should be assigned depending on the definitive diagnosis, fertility potential, genital appearance, surgical options, and the parents’ opinion. For instance, in 46, XX CAH, which is the most common type of DSD, and virilization due to maternal androgen, there is high fertility potential.16 Almost all patients with 46, XX CAH and CAIS are raised female.17,18 During the evaluation of normal sexual function capacity in PAIS, supra-physiological doses of testosterone can be attempted for the measurement of penis/phallus size. Most of the patients with enzyme deficiencies of 5-α reductase-2 and 17 β-hydroxysteroid (40–60%) are first accepted as female, while they maintain their lives as men after puberty.19 The presence of the distal vagina opening is a facilitating factor for the adoption of the female identity, while a proximal high vagina opening can result in difficulty in repair, and the chances of fertility must be considered in these cases. Although mild clitoromegaly is considered a flaw, it can sometimes lead to a very colorful sex life.
When deciding on the patient's gender, gonadal status and hormone levels should be considered. In 5-α reductase enzyme deficiency, during the identification in the infancy, male gender identity and fertility should be considered when making the decision.19,20 It should be noted that 46, XX virilized females have functional ovaries. In the ovotesticular DSD patients, who have both ovaries and testicles, gender determination should be assigned according to the gonadal development, taking hormonal functionality into account. In ovotesticular DSD and mixed gonadal dysgenesis (MGD) patients (usually 45 XO/46 XY mosaic), gonadal function and phallic appearance should be evaluated and it should be kept in mind that the testicles are functional but insufficient.
In CAIS there is no effect of androgen due to a defect in androgen receptors. Accordingly, the patients have a female appearance and they tend to be raised as female.21 Patients with incomplete gonadal dysgenesis and PAIS are in the most difficult group for gender assignment, because 25% of these patients will be unhappy no matter in which gender they are raised.22 On the other hand, 50% of men with normal sexual development and 20% of women can have a satisfying sex life, while most patients with congenital micropenis have quite enjoyable sexual experiences.23 The patients with 46, XY cloacal exstrophy behave like males even when they are raised as females, and some of them do return to the male identity.24,25
Medical treatment of DSD46, XX CAH is the most common cause of ambiguous genitalia in children, with an autosomal recessive inheritance. It may create adrenal crisis due to loss of salt, which is an emergency. CAH is caused by dysfunction of one of the five enzymes responsible for the synthesis of cortisol. Of all the cases, 90–95% has a deficiency of the 21-hydroxylase enzyme. The second most common enzyme deficiency is that of 11-beta-hydroxylase enzyme.26–28 CAH can be classified as clinical, hormonal and molecular types. Clinically, it can be divided into, being severe, classical and mild, non-classical types. The patients with classical type of CAH are further classified as simple and virilizing depending on the degree of aldosterone deficiency.5
In neonates with CAH, the treatment consists of glucocorticoid and mineralocorticoid replacement. The goal of treatment is to attenuate the cortisol deficiency, and to keep the androgen at normal levels depending on the age.
In DSD, the basis of medical treatment consists of the replacement of the missing hormones. The most obvious treatment applications are cortisol treatment in adrenal hyperplasia, and testosterone therapy in children with androgen biosynthetic defects and who are raised as males, in order to facilitate the growth of the phallus and puberty. In girls, different forms of estradiol can be applied in daily doses or in depot forms. Along with the replacement therapy, bone development should not be neglected, and must be supported, as well.
Surgical treatment of DSDWhat can be accomplished through surgery primarily depends on the anatomic structure of the child. In patients with adrenogenital syndrome who appear to be male while they are female, the patients who are male pseudo-hermaphrodite with underdevelopment of male external genitalia or the patients with MGD, and some of the true hermaphrodites, the perineum should be reconstructed as female. If possible, the repair should be done between 3 and 6 months after birth. An important step is to reduce the size of the clitoris. In cases with adrenogenital syndrome, the vagina frequently opens to the urogenital sinus distally to the external urethral sphincter, for which a simple cut-back or flap vaginoplasty is sufficient. Vaginoplasty can be performed in the same session as the clitoroplasty. During the clitoroplasty, the neurovascular package in the dorsal aspect of the phallus must be protected. If the vagina opens to the urogenital sinus proximally to the external urethral sphincter, clitoroplasty is completed, and vaginoplasty can be postponed until the patient is two years old. In cases of male pseudo-hermaphrodite, the phallus should be minimized without delay and subsequently vaginoplasty should be performed before puberty. Using sigmoid colon to create a vagina during childhood is the most suitable option. In real hermaphrodites, the gonads are preserved while the external genital structures are reconstructed as female.
DSD patients are at risk of developing germ-cell tumors and gonadoblastoma. The risk is very high in patients with gonadal dysgenesis and PAIS with intra-abdominal gonads, whereas patients with ovotesticular DSD have a lower risk (<5%).29 Nowadays, prophylactic gonadectomy is postponed until puberty in such CAIS cases to maintain the hormonal balance and due to the low risk of malignancy on contrary to considered.1 On the other hand, streak gonads should be removed early due to the high risk of malignancy.
ConclusionDisorders of sex development are the complex cases. Evaluation and management of this patients group must be organized as quickly as possible and accurately with a careful multidisciplinary team. The immediate interventions should be done timely and in an accurate manner. The gender assignment is decided according to the definitive diagnosis, fertility potential, genital appearance, surgical options, and the parents’ opinion.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe author declare no conflict of interest.