


INTRODUCCIÓN
La fibrosis quística (FQ), cuyo nombre se refiere a los procesos característicos de cicatrización (fibrosis) y formación de quistes dentro del páncreas, reconocidos por primera vez en los años treinta del pasado siglo, también conocida como mucoviscidosis (del latín muccus, "moco", y viscÿsus, "pegajoso"), es una enfermedad hereditaria frecuente que afecta al organismo de forma generalizada, causando discapacidad progresiva y muerte prematura. Se trata pues de una de las enfermedades hereditarias fatales más comunes. Su prevalencia es mayor entre caucásicos, variando su incidencia de 1 entre 2.500 a 1 entre 8.000 nacidos vivos1,2. Una de cada 25 personas de ascendencia europea es portadora asintomática de un gen para FQ, siendo la enfermedad genética más frecuente entre esta población. Los afectados pueden ser diagnosticados mediante pruebas genéticas prenatales; también por cribado neonatal o, durante la infancia temprana, por la mencionada prueba del sudor3. La FQ no tiene cura, y la supervivencia media para estos pacientes se estima en 29 años, alcanzando valores más altos en algunos países, como por ejemplo en Francia que es de 35 años4. En casos severos, la mala evolución de la enfermedad puede imponer la necesidad de un trasplante de pulmón.
La FQ está causada por una mutación en un gen llamado regulador de la conductancia transmembrana de la fibrosis quística (CFTR). El gen está situado en el cromosoma 7 y se aisló en 19895-7. En este gen se han descrito más de 1.000 mutaciones asociadas a la enfermedad. De estas mutaciones, la más frecuente es la ΔF 508 y se produce por la pérdida del aminoácido fenilalanina en la posición 508. Esta mutación representa el 70% de las mutaciones en los europeos de origen caucásico. La naturaleza de la mutación se correlaciona con la gravedad de la alteración pancreática y el grado de anormalidad de cloro en el sudor, y también está asociada con disminución de la fertilidad en ambos sexos. Este gen interviene en la producción de sudor, jugos gástricos y moco. Aunque la mayoría de las personas sanas tienen 2 copias funcionales del gen, sólo 1 es necesaria para impedir el desarrollo de fibrosis quística. La FQ se presenta cuando ninguno de estos genes opera normalmente. En consecuencia, se la considera una enfermedad auto-sómica recesiva.
Se ha demostrado que hay una mayor prevalencia de mutaciones del gen CFTR en varones saludables, con una reducida calidad espermática, que en varones controles, por lo que en los pacientes subfértiles el riesgo de tener una descendencia afectada de FQ es superior a los que tienen una calidad seminal normal8,9.
Dada la alta incidencia de esta enfermedad en nuestra población y los beneficios que nos ofrece el diagnóstico genético preimplantacional (DGPI), para evitar las consecuencias desastrosas que podrían presentar personas nacidas de embriones afectados de esta patología, nos propusimos publicar este caso clínico, recalcando que ambos miembros de la pareja son portadores del gen de la FQ.
CASO CLÍNICO
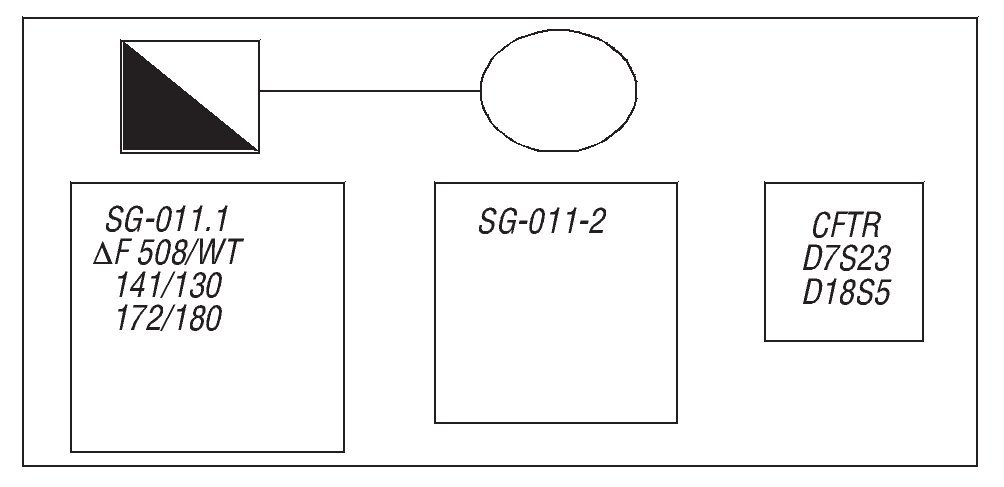
Pacientes JLB y MLSA, que son estudiados por su médico de asistencia por una infertilidad primaria. En el varón se encuentra una oligoazoospermia severa en el espermiograma, por lo que se somete a las pruebas de cribado previas a la realización de fecundación in vitro (FIV)/inyección intracitoplasmática de espermatozoides (ICSI) (gen DAZ, FQ y cariotipo), y se le diagnostica como portador del gen ΔF 508 de FQ; secundariamente se estudia a la mujer, la cual aparece como portadora de los genes ΔF 508/L206W de FQ (fig. 1), además de presentar antecedentes de una ooforectomía unilateral anterior por un cistoadenoma benigno del ovario. Con estos antecedentes, los pacientes son enviados para realización de FIV/ICSI.
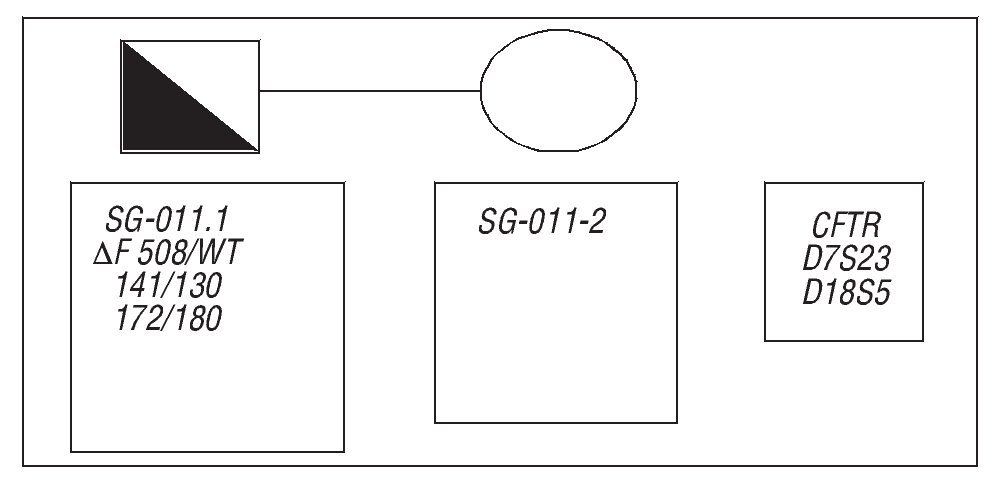
Figura 1. Mutaciones del gen de la fibrosis quistica en ambos miembros de la pareja.
Dicha FIV, en la modalidad de ICSI, se realiza bajo estimulación de la ovulación con antagonistas (Cetro-tide®) y gonadotropinas recombinantes (Puregon®). Cuando 4 folículos presentaron un diámetro ≥ 18 mm, se administró 10.000 unidades de gonadotropina coriónica humana (HCG). La captación ovocitaria se realizó a las 35 h y media, bajo sedación con propofol.
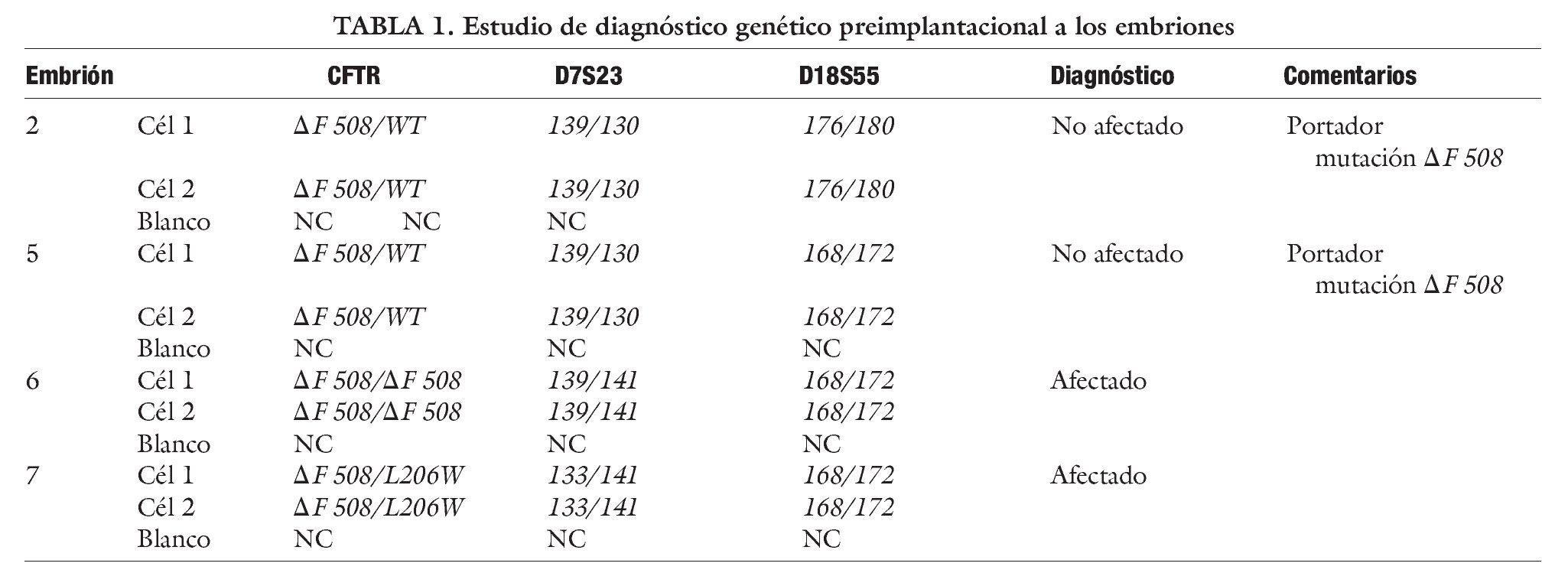
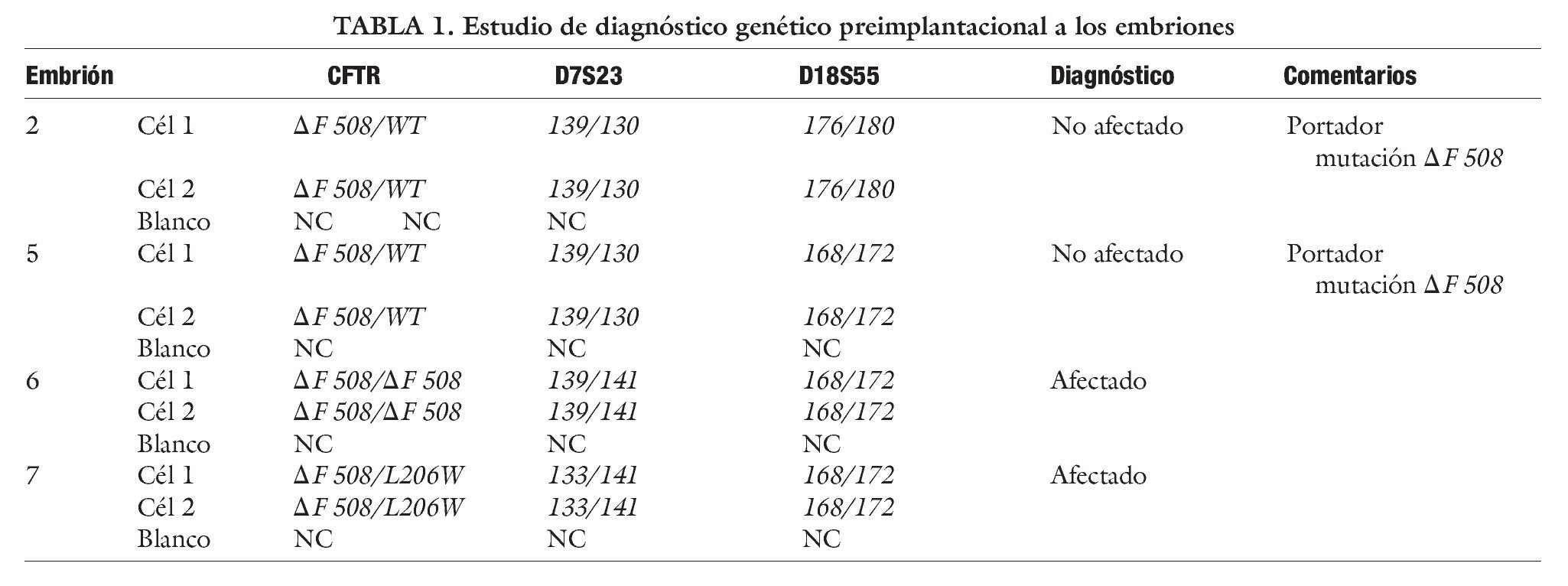
Se obtuvieron 15 ovocitos, de ellos 13 MII y se lograron 12 embriones en el día +2. El día después de la punción la paciente acudió a urgencias de nuestra clínica con dolor abdominal y malestar general. Se constató sangrado abdominal pospunción, por lo que se somete a laparoscopia urgente con resultado favorable. Se decide congelar todos los embriones el día +2 y posponer el DGPI para un ciclo posterior con los embriones congelados.
Tres meses después y en ciclo espontáneo se descongelaron los 12 embriones y sólo 4 sobrevivieron el día +3, a los cuales se realizó la biopsia embrionaria sin incidencias. El día +4 se informó que 2 de los embriones eran portadores y otros 2 afectados (tabla 1). Se realizó la transferencia de los 2 embriones portadores el día +5, que estaban en estadio de blastocistos. Se suplementó el ciclo con progesterona micronizada y acido fólico. El día 11 postranferencia, se verificó embarazo por βHCG en sangre y 2 semanas más tarde se confirmó por ecografía la presencia de un saco gestacional con embrión. El embarazo evolucionó sin incidencias finalizando en un parto por cesárea de niña sana, con un peso de 3.200 g y apgar 9-10, portadora de la mutación ΔF 508 de la FQ.
DISCUSIÓN
Este caso nos reafirma la necesidad de un estudio a fondo de las causas que pueden llevar a infertilidad y de las posibles asociaciones entre infertilidad y anomalías génicas. Además de en la FQ10-12, objetivo de esta publicación y que por su incidencia en nuestra población es de mucha importancia, hay numerosas patologías donde la aplicación del DGPI a embriones, obtenidos por FIV, toma utilidad como forma temprana de diagnóstico prenatal, como ocurre en la enfermedad de Huntington11,12, hemofilia A13 y distrofia miotonica12,14, entre otras.
CONCLUSIÓN
El DGPI ha demostrado ser una herramienta fundamental, no sólo para la obtención de la gestación sino también para lograr el nacimiento de niños sanos.
Correspondencia:
Dr. M. Brassesco.
Centro de Infertilidad y Reproducción Humana (CIRH).
Plaza Eguilaz, 14, bajo. 08017 Barcelona. España.
Correo electrónico: cirh@cirh.es