


INTRODUÇÃO
Os estados intersexuais são situações raras com uma prevalência de 1 para 25000 nados vivos na Europa. Nas situações de ambiguidade sexual não é possível caracterizar ao nascer, um indivíduo como sendo do sexo masculino ou feminino, baseado apenas no exame físico, pois coexistem nesse mesmo indivíduo elementos anatómicos com características de ambos os sexos.
A classificação destes estados intersexuais embora complexa fundamenta-se no cariótipo e na presença de gónadas. Habitualmente divide-se em cinco tipos: hermafroditismo verdadeiro, pseudohermafroditismo feminino, pseudohermafroditismo masculino, disgenesia gonadal pura e disgenesia gonadal mista.
O termo pseudohermafroditismo masculino refere-se a indivíduos 46XY, que apesar da presença de testículos, apresentam diferentes graus de fenótipo feminino. A deficiente diferenciação masculina destes indivíduos pode ser devida à inadequada produção de testosterona, a uma insensibilidade parcial dos tecidos aos androgénios ou a uma deficiente produção ou acção da substância inibidora mulleriana (MIS).
Durante a vida fetal é necessária a isoenzima 17β-hidroxiesteroide dehidrogenase tipo 3 (17β-HSD-3) para a síntese da testosterona (T). Esta, tem de ser convertida em dihidrotestosterona (DHT), o seu metabolito activo nos receptores dos tecidos periféricos, e que vai permitir o desenvolvimento dos genitais masculinos externos. A deficiência desta 17β-hidroxiesteroide dehidrogenase (17β-HSD) trata-se duma forma de pseudohermafroditismo, com transmissão autossómica recessiva devido à deficiência na conversão de androstenediona (A) em testosterona.. Esta enzima é responsável pelo último passo da síntese dos androgénios - a conversão de androstenediona em testosterona, dihidroepiandrostenediona em androstenediol e estrona em estradiol. Este quadro foi descrito pela primeira vez, em 1971, por Saez.
Esta situação é, clinicamente, muito semelhante à deficiência de 5_-redutase, na qual os indivíduos afectados, também apresentam os testículos localizados no canal inguinal ou na bolsa labio-escrotal. O fenótipo pode variar desde ambiguidade genital ao nascer até a uma quase feminilização completa dos genitais externos, sendo educados como se fossem do sexo feminino. No entanto, estes indivíduos possuem testículos bem diferenciados, derivados dos canais de Wolf hipoplásicos e uma ausência dos derivados dos canais de Muller. Como as gónadas não são removidas, inicia-se uma virilização durante a puberdade, com o crescimento do pénis e o desenvolvimento de características sexuais secundárias masculinas, como seja o aumento da massa muscular com distribuição masculina, o desenvolvimento do pêlo púbico, axilar, da face e corpo. Esta virilização tardia parece ser devida à produção de testosterona por outras isoenzimas 17β-HSD.
MATERIAL E MÉTODOS
Luzia C, 17 anos, solteira, raça negra, natural de Angola. A criança foi criada como rapariga até à puberdade, altura em que iniciou uma virilização progressiva, motivo pelo qual foi enviada à consulta de Urologia do Hospital Geral de Santo António.
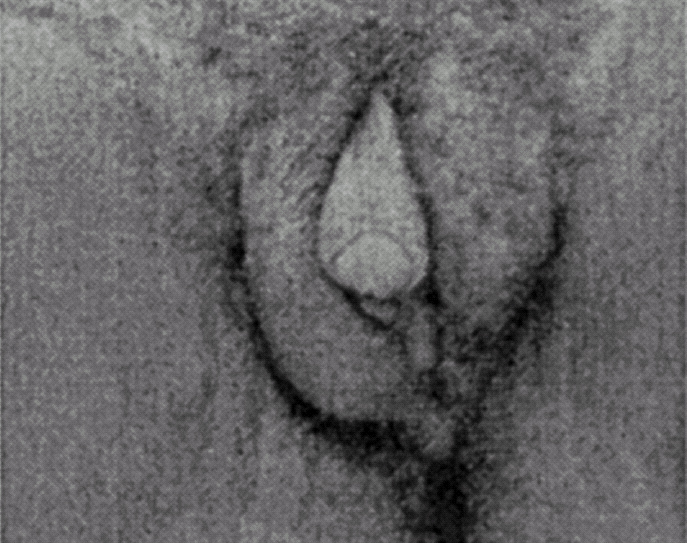
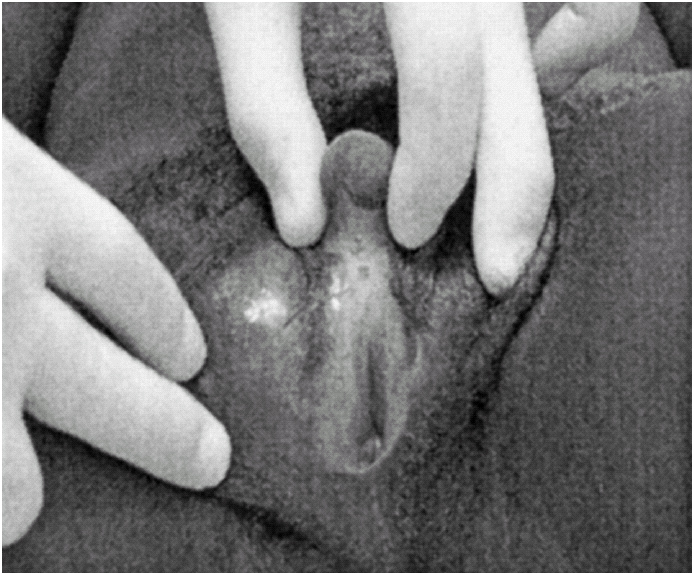
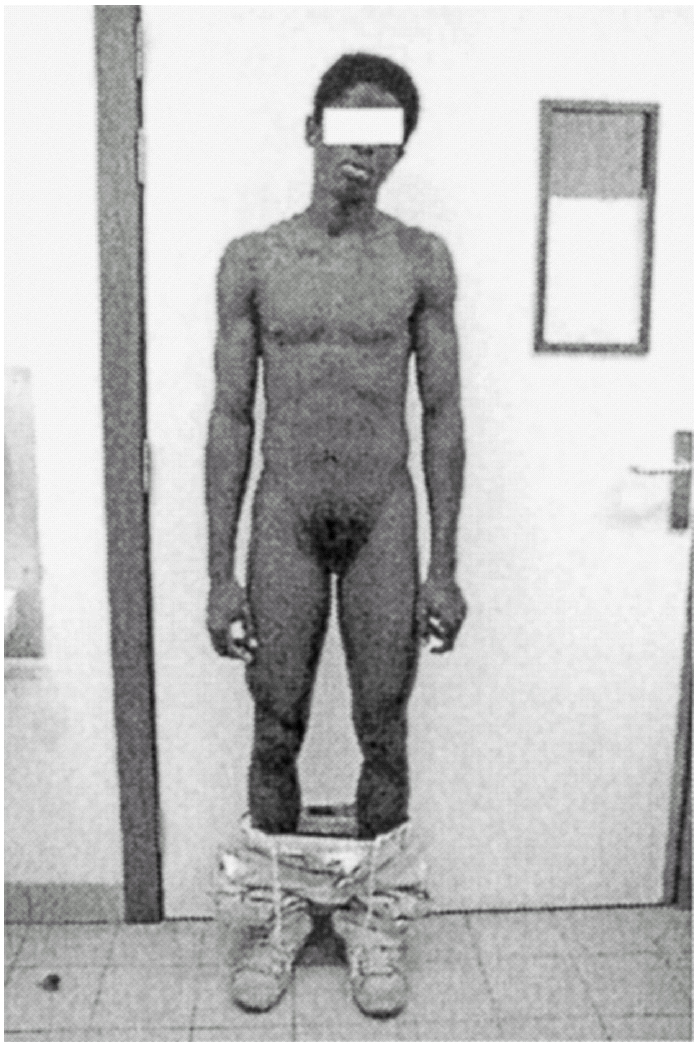
Ao exame físico, apresentava fenótipo masculino, de voz grossa, com uma massa muscular desenvolvida, de tipo atlético, testículos palpáveis nas bolsas lábio-escrotais (fig. 1), uma ambiguidade genital com pénis clitoriforme e vagina cega (fig. 2) a ausência de ginecomastia, e uma rarefacção pilosa para o sexo e idade (fig. 3).
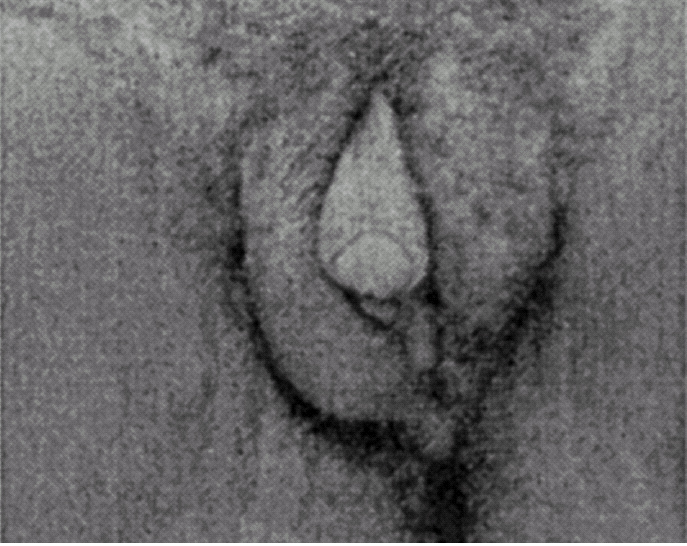
Figura 1.
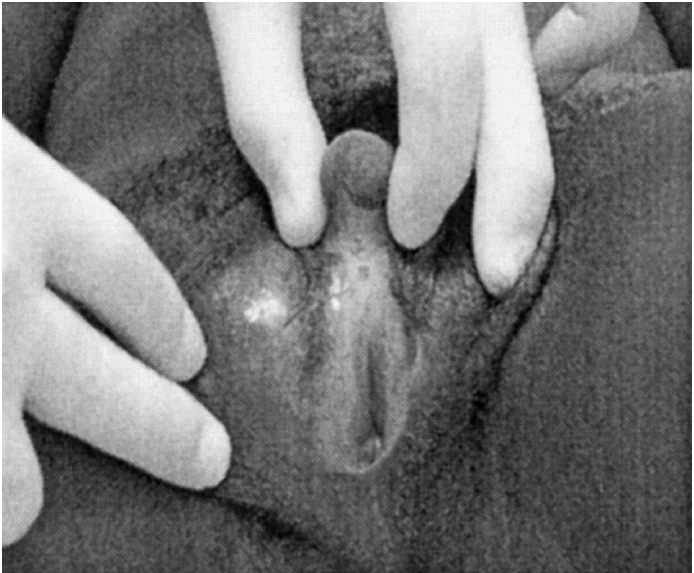
Figura 2.
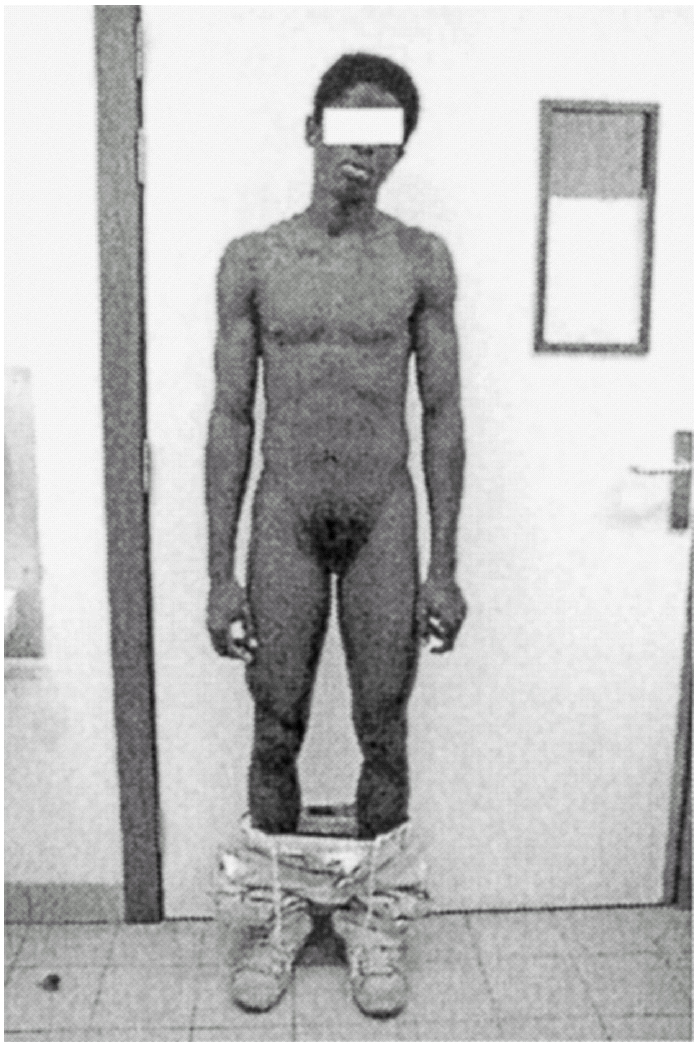
Figura 3.
O estudo genético revelou um cariótipo 46XY.
O estudo ecográfico revelou ambos os rins e a bexiga sem alterações, a próstata e vesículas seminais, atrofiadas, de aspecto rudimentar. O testículo e epidídimo esquerdos, embora com aspecto normal, encontravam-se localizados na bolsa lábio-escrotal homolateral. O testículo direito também com uma localização lábio-escrotal, mas tinha dimensões reduzidas e uma ecoestrutura heterogénea. O epidídimo direito não foi visualizado.
O estudo analítico apresentava hemograma, a função renal e hepática sem alterações. Foi efectuada a medição dos valores de Testosterona e Androstenediona antes e após estimulação com β-HCG, a qual revelou um ratio de A / T> 1, e suficiente por si só para permitir o diagnóstico de deficiência de 17β-HSD.
A avaliação psicológica e psiquiátrica/sexológica consistiram na entrevista semi-estruturada, com dados baseados na clínica e o recurso a meios de avaliação estandardizados. Estes, têm por finalidade qualificar o diagnóstico psíquico, a identidade e orientação sexual, o funcionamento sexual, a imagem corporal, o papel sexual, o impacto social bem como o índice de masculinidade/feminilidade. Os dados registados indicavam um doente com identidade e orientação não definida. Iniciou psicoterapia e após reatribuição de género foi então submetido a faloplastia e correcção estética do escroto em Setembro de 2004.
CONCLUSÃO
Os estados intersexuais são situações raras. O seu diagnóstico deverá ser efectuado à nascença, altura ideal para iniciar o tratamento e orientação correctas (atribuição de género e substituição hormonal/gonadectomia). Neste caso clinico, o diagnóstico foi efectuado muito tardiamente, apenas na altura da puberdade, pelo que se optou por proceder a faloplastia e correcção dos genitais externos, no sentido masculino, após a reatribuição de género.
Correspondencia: R. Borges.
Serviço de Urologia do HGSA. Porto.
Bibliografia
David A, Diamond MD, Sexual differentiation: normal and abnormal. Campbell's Urology, chapter 68.
Dennis M. Styne: The testes: Disorders of sexual differentiation and puberty. Pediatric Endocrinology, chapter 16.
Melvin M. Grumbach, Ieuan A. Hughes, Felix A. Conte: Disorders of sex differentiation. Williams Textbook of Endocrinology.