













En este trabajo se emplean métodos de inteligencia computacional, tales como las máquinas de soporte vectorial (MSV) para optimizar la predicción de la solubilidad de compuestos. Estas se entrenan con una base de datos de compuestos solubles e insolubles conocidos, y dicha información es posteriormente empleada para mejorar la predicción obtenida mediante cribado virtual. Los grandes avances en el campo de la computación de alto rendimiento ofrecen nuevas oportunidades en la simulación de sistemas biológicos y aplicaciones en bioinformática, biología computacional y química computacional. El uso de bases de datos de mayor tamaño aumenta las posibilidades en la generación de candidatos potenciales, pero el tiempo de cálculo necesario no sólo aumenta con el tamaño de la base de datos, sino también con la exactitud de los métodos de cribado virtual (CV) y del modelo. Se discuten los beneficios del uso de arquitecturas masivamente paralelas, en particular las unidades de procesamientos gráfico, demostrando empíricamente que están bien adaptadas para la aceleración de las MSV, obteniendo una aceleración de hasta 45 veces, en comparación con su versión secuencial.
In this work we discuss the benefits of using computational intelligence methods, like Support Vector Machines (SVM) for the optimization of the prediction of compounds solubility. SVMs are trained with a database of known soluble and insoluble compounds, and this information is being exploited afterwards to improve Virtual Screening (VS) prediction. The landscape in the high performance computing arena opens up great opportunities in the simulation of relevant biological systems and for applications in bioinformatics, computational biology and computational chemistry. Larger databases increase the chances of generating hits or leads, but the computational time needed for the calculations increases not only with the size of the database but also with the accuracy of the VS methods and the model. We discussed the benefits of using massively parallel architectures, in particular graphics processing units. We empirically demonstrate that GPUs are well-suited architecture for the acceleration of SVM, obtaining up to 15 times sustained speedup compared to its sequential counterpart version.
El descubrimiento de nuevos fármacos es un proceso complicado que puede ser optimizado, en las primeras etapas, gracias a la utilización de métodos de cribado virtual (CV o Virtual Screening, VS en inglés). Las limitaciones de las predicciones del CV están directamente relacionadas con la falta de recursos computacionales, un importante cuello de botella que impide la aplicación de modelos detallados de alta precisión al CV.
En éste trabajo, nos centramos en la optimización del cálculo de la predicción de la solubilidad de compuestos usando métodos de inteligencia computacional cómo las Máquinas de Soporte Vectorial (MSV o Support Vector Machines, SVM en inglés). Las MSV son entrenadas con una base de datos de compuestos solubles e insolubles conocidos, y esta información es posteriormente usada para mejorar la predicción del CV.
La complejidad de los descriptores empleados en la predicción, el gran tamaño de las bases de datos empleadas y la naturaleza intrínsecamente paralela de los métodos de inteligencia computacional empleados sugieren que para obtener resultados significativos se explote dicho paralelismo. De entre los recursos de computación que emplean esta estrategia, el uso de arquitecturas masivamente paralelas, unidades de procesamiento gráfico (GPU del inglés Graphic Processor Units), ofrece un gran rendimiento sobre una amplia variedad de aplicaciones y, sobre todo, en este tipo de métodos de simulación [1].
Las nuevas generaciones de GPUs son procesadores masivamente paralelos que pueden soportar varios miles de subprocesos simultáneos. Las tarjetas NVIDIA actuales [2] contienen multitud de elementos de procesamiento y se programan utilizando una extensión del lenguaje C llamado CUDA (del inglés Compute Unified Device Architecture) [3]. En las GPUs, la aceleración alcanza en muchos casos hasta 100 veces [4,5], respecto a versiones secuenciales sobre CPU. Es tal la eficiencia de estos dispositivos, que en la arquitectura NVIDIA Kepler algunos modelos de GPU alcanzaron un rendimiento máximo por encima de 3.52 TFLOPS [6].
El resto del trabajo se organiza de la siguiente manera. La Sección 2 presenta la metodología incluyendo los fundamentos de las MSV, las bases de datos de ligandos y propiedades moleculares empleadas, la descripción de las arquitecturas GPU y el modelo de programación CUDA de NVIDIA. La Sección 3 describe las implementaciones secuencial con CPU y paralela sobre GPU de las MSV. La evaluación del rendimiento se discute en la Sección 4. Finalmente, la Sección 5 describe algunas conclusiones e ideas para trabajos futuros.
2MetodologíaEn esta sección se describen los métodos utilizados para mejorar la predicción de la solubilidad. Una técnica de Inteligencia Computacional cómo las MSV se emplea para la predicción de la solubilidad y para ello este método se entrenó con diferentes conjuntos de datos. También se muestra la arquitectura GPU utilizada para acelerar el proceso, y el conjunto de datos y descriptores moleculares utilizados.
2.1Máquinas de Soporte VectorialLas máquinas de vectores soporte vectorial (MSV) [8] son un grupo de métodos de aprendizaje supervisado que pueden ser aplicados a problemas de clasificación o regresión. Fueron inicialmente introducidos para clasificar clases de objetos linealmente separables. Representan hiperplanos o fronteras de decisión en términos de un pequeño subconjunto de todos los ejemplos de entrenamiento que maximizan la separación entre conjuntos y por lo tanto la capacidad de clasificación entre clases, denominados vectores de soporte. En un corto período de tiempo, las MSV han sido aplicadas en numerosas aplicaciones de química, tales como el diseño de fármacos (discriminación entre ligandos y no ligandos, inhibidores y no inhibidores, etc.) [9], el descubrimiento de fármacos [10], las relaciones cuantitativas estructura-actividad (QSAR), donde se utiliza la regresión MSV para predecir diversas propiedades biológicas, físicas y químicas [11], la quimiometría (optimización de la separación cromatográfica o predicción de la concentración del compuesto a partir de datos espectrales) [12], sensores (para la predicción cualitativa y cuantitativa de datos) [13], o ingeniería química (detección de fallos y la modelización de procesos industriales) [14]. Una excelente revisión de las aplicaciones de MSV en química fue publicada por Ivancicuc [15].
En nuestro caso, explotamos la idea de que las MSV producen un hiperplano particular en el espacio de características que separa los compuestos solubles o insolubles, al que llamamos hiperplano de máximo margen (ver fig. 1). Sin embargo las funciones base para definir los vectores de soporte que definen los hiperplanos pueden ser de varios tipos: lineal, polinómico, neuronal (sigmoide, Tanh), Anova, Fourier, Spline, B Spline, aditivo, tensor y Gaussiana de base radial o exponencial.
2.2Base de datos de ligandos y propiedades moleculares
Empleamos conjuntos de prueba estándar en CV, tales como NCI (Versión 4 - mayo 2012) que es una gran base de datos de moléculas [16]. A continuación, utilizando el paquete ChemoPy [17], se calculó, para todos los conjuntos de ligandos, un subconjunto diverso de propiedades moleculares derivadas del conjunto de descriptores de Constitución, CPSA (carga parcial y área de superficie) y descriptores basados en huellas o fragmentos, como se describe en la tabla 1.
Descriptores moleculares usados en el estudio
| DESCRIPTORES CONSTITUCIONALES | |
| Natom | Número de átomos |
| MolWe | Peso Molecular |
| NRing | Número de anillos |
| NArRg | Número de anillos aromáticos |
| NRotB | Número de enlaces rotativos |
| NHDon | Número de H-enlaces donantes |
| NHAcc | Número de H-enlaces aceptores |
| DESCRIPTORES CPSA | |
| Msurf | Área de superficie molecular |
| Mpola | Área polar de superficie molecular |
| Msolu | Solubilidad molecular |
| AlogP | Coeficiente de partición |
| DESCRIPTORES BASADOS EN FRAGMENTO/HUELLA | |
| ECP2,ECP4,ECP6 | Huellas de conectividad extendida (ECFP) |
| EstCt | Contador de estados |
| AlCnt | AlogP2 Contador de estados |
| EstKy | Clave de estados |
| MDLPK | Claves públicas de descriptor molecular |
Las propiedades constitucionales dependen de descriptores muy simples de la molécula que se pueden calcular fácilmente, simplemente contando el número de elementos moleculares, tales como átomos, tipos de átomos, enlaces, anillos, etc. Estos descriptores deben ser capaces de diferenciar moléculas muy diferentes, pero podrían tener problemas para separar isómeros estrechamente relacionados. Los descriptores CPSA consideran detalles más finos de la estructura molecular por lo que son capaces de separar las moléculas similares, pero también pueden tener dificultades para separar isómeros. Por último, los descriptores de fragmento y los basados en huellas consideran la presencia de una estructura exacta (no una subestructura) con un número limitado de puntos de fijación especificados. En la generación de huellas, el programa asigna un código inicial a cada átomo. El código inicial de átomo deriva del número de conexiones de este, el tipo de elemento, carga atómica, y la masa atómica. En este caso corresponde a una huella de conectividad extendida (en inglés ECFP) con un tamaño de la vecindad cero. Estos códigos de átomos se actualizan de forma iterativa para reflejar los códigos de cada átomo vecino. En la siguiente iteración, se emplea un esquema de cifrado para incorporar información de cada átomo vecino inmediato. De esta forma, cada nuevo código átomo describe una estructura molecular con un tamaño de la vecindad de uno. Este proceso se lleva a cabo para todos los átomos de la molécula. Cuando se alcanza el tamaño de vecindad deseado, el proceso se ha completado y el conjunto de todas las características se devuelve como la huella. Para las ECFPs empleados en este trabajo, tamaños de vecindad de dos, cuatro y seis (ECFP 2, ECFP 4, ECFP 6) se utilizaron para generar las huellas. Las ECFPs resultantes pueden representar un conjunto mucho mayor de características que otras huellas y contienen un número significativo de diferentes unidades estructurales, crucial para la comparación molecular entre los compuestos.
Con el fin de encontrar la mejor opción para discriminar entre compuestos solubles e insolubles en estos conjuntos de datos, utilizaremos descriptores basados en huellas y evitaremos el uso de descriptores constitucionales y CPSA. Esto es razonable, ya que los descriptores de huellas consideran más detalles acerca de la estructura de las moléculas, al ser capaces de discriminar de manera eficiente y con una mayor precisión entre los compuestos activos y sus señuelos.
A continuación, estudiamos qué combinación de propiedades podría conducir a mejoras en la capacidad de predicción de estos métodos de computación flexible; es evidente que con estas combinaciones la capacidad de predicción aumentará (véase la tabla 2).
Combinaciones de descriptores moleculares usados en el estudio
| COMBINACIONES DE DESCRIPTORES CONSTITUCIONALES | |
| MNBH | Área Polar de superficie molecular (MPola)+ Número de enlaces rotables (NRotB) + Número de H-enlaces aceptores (NHAcc) |
| MNB | Área Polar de superficie molecular (MPola) + Número de enlaces rotables (NRotB) |
| NBH | Número de enlaces rotables (NRotB) + Número de H-enlaces aceptores (NHAcc) |
| MoN | Área Polar de superficie molecular (MPola) + Número de H-enlaces aceptores (NHAcc) |
| MoN | Área Polar de superficie molecular (MPola) + Número de H-enlaces aceptores (NHAcc) |
| COMBINACIONES DE DESCRIPTORES BASADOS EN FRAGMENTO/HUELLA | |
| EAE246 | Contador de estados (EstCt) + AlogP2 Contador de estados (AlCnt) + Huella de conectividad extendida (ECFP) |
| EA | Contador de estados (EstCt) + AlogP2 Contador de estados (AlCnt) |
| AE246 | AlogP2 Contador de estados (AlCnt) + Huella de conectividad extendida (ECFP) |
| EE246 | Contador de estados (EstCt) + Huella de conectividad extendida (ECFP) |
Como mencionamos anteriormente, el proceso de aplicar los métodos de inteligencia computacional para predecir la solubilidad en grandes bases de datos de elementos consume mucho tiempo. Para acelerar éste proceso, proponemos el uso de hardware de alto rendimiento. Las GPUs son dispositivos compuestos por un gran número de unidades de proceso, que en un principio fueron diseñados para ayudar a la CPU en los cálculos gráficos, pero han evolucionado en el transcurso del tiempo para convertirse en dispositivos programables capaces de obtener una gran capacidad de cómputo tanto en aritmética entera como de punto flotante.
Cuando se programan correctamente, las GPU se pueden utilizar cómo procesadores de propósito general. El código usado para los procesadores secuenciales necesita ser readaptado para estas arquitecturas, y esto no es siempre un proceso sencillo. Durante los últimos años, las mayores facilidades de programación y la introducción de nuevas herramientas y lenguajes ha permitido la creación de una comunidad de investigación más amplia para explotar la programación de la GPU.
Las GPU son dispositivos altamente paralelos con cientos o miles de núcleos. Funciones matemáticas especiales, frecuentemente empleadas en los cálculos científicos cómo operaciones trigonométricas o raíces cuadradas, se implementan directamente sobre el hardware, lo que permite realizar muy rápidamente dichas operaciones. El rendimiento teórico máximo varía entre los diferentes modelos de GPU. La arquitectura de la GPU NVIDIA se basa en conjuntos de procesadores escalares (SP) organizados como multiprocesadores (SM) que comparten una memoria con una latencia de acceso muy baja (fig. 2).

El modelo de programación CUDA permite codificar programas paralelos para las GPU utilizando algunas extensiones del lenguaje C. Un programa de CUDA se divide en dos partes principales: el programa que se ejecuta en la CPU (parte del host) y el programa ejecutado en la GPU (parte del dispositivo), que se llama kernel. En el kernel hay dos niveles principales de paralelismo: hilos CUDA, y bloques de hilos CUDA [7].
3Implementación de las Máquinas de Soporte VectorialEn esta sección describimos la experimentación de MSV sobre el paquete de R [18] para ambas versiones: CPU y GPU. Vamos a comparar la experimentación de la versión secuencial de MSV con la versión paralela en la predicción de la solubilidad, inicialmente con un solo descriptor molecular sencillo (ALOGP) y posteriormente con un descriptor molecular complejo (EAE246).
3.1Versión Secuencial (CPU)El paquete e1071 se basa en la biblioteca LIBMSV [19] que se está utilizando en algunos métodos eficientes de selección automática de parámetros. Es un paquete secuencial que hace uso de la CPU.
En R están disponibles diferentes implementaciones de MSV. Con el fin de validar los resultados, hemos elegido este paquete, ya que obtiene buenos resultados en los conjuntos de datos estándar. En la tabla 3 se comparan las cuatro implementaciones MSV [20] en términos de tiempo de entrenamiento. Esta comparación se centra en el tiempo de entrenamiento de MSV, excluyendo el tiempo necesario para la estimación del error de entrenamiento o el error de validación cruzada. En estas implementaciones se utilizó el paquete de MSV en R. El conjunto de datos utilizado es el conjunto de datos estándar del UCI Repository of Machine Learning Databases [21].
Tiempos de entrenamiento en segundos para las implementaciones secuenciales sobre diferentes conjuntos de datos en segundos. Se empleó un computador Intel Core I3 3.07 GHz
| Datos | Kernlab | E1071 | klaR | MSVpath |
|---|---|---|---|---|
| Spam | 18.50 | 17.90 | 34.80 | 34.00 |
| Musk | 1.40 | 1.30 | 4.65 | 13.80 |
| Vowel | 1.30 | 0.30 | 21.46 | NA |
| DNA | 22.40 | 23.30 | 116.30 | NA |
| BreastCancer | 0.47 | 0.36 | 1.32 | 11.55 |
| HouseBoston | 0.72 | 0.41 | 92.30 | NA |
La versión paralela de MSV utilizada es parte del paquete RPUDPRO [22], que se ha construido en base a una versión paralela de MSVLIB. Para procesar la versión paralela de MSV, consideramos que el problema de la clasificación con MSV se puede separar en dos tareas diferentes; el cálculo de la matriz del núcleo o función (KM) y la tarea esencial en MSV de descomponer y resolver el modelo de clasificación. El tamaño creciente de los datos de entrada conduce a una enorme KM que no puede ser calculada y almacenada en la memoria (ver Algoritmo 1). Por lo tanto, el procedimiento necesita ir calculando porciones de KM, lo que requiere de procesamiento y uso intensivo de ancho de banda de memoria. LIBMSV utiliza doble precisión para los cálculos, pero sólo las últimas GPUs soportan doble precisión. El precálculo se realiza combinando la CPU y la GPU para lograr el máximo rendimiento.
Algoritmo 1. Pseudocódigo Paralelo
| 1: Precálculo en la CPU los elementos para cada vector de entrenamiento. |
| 2: Convertir la matriz en columnas vectores de entrenamiento. |
| 3: Asignar en la memoria del dispositivo de la GPU la matriz vectores de entrenamiento. |
| 4: Cargar la matriz vectores de entrenamiento a la memoria de la GPU. |
| 5: PARA (cada vector de entrenamiento) HACER |
| Cargue el vector de la formación a la GPU. |
| Realizar operaciones con CUBLAS. Recuperar el producto escalar del vector de la GPU. FIN HACER 6: Desasignar la memoria de la GPU |
Como puede verse en el pseudocódigo del algoritmo 1, el algoritmo propuesto se basa en precalcular en la CPU, para cada cálculo del vector de entrenamiento, usando CUBLAS [17] biblioteca proporciona desde NVIDIA
4ExperimentaciónLa experimentación realizada pretende contrastar tanto la capacidad predictiva del método como la conveniencia de la paralelización y aceleración sobre procesadores gráficos. Así como asegurar el mantenimiento de las capacidades predictivas del método al reconvertir el algoritmo para su adaptación a la arquitectura GPU.
La base de datos empleada es Open NCI Database [21] que se hizo pública en 1999. Se compone de 249 000 estructuras moleculares.
El rendimiento de las implementaciones secuenciales y GPU se evalúa sobre un computador Intel Core I3 540 a 3.07GHz y 8GB de memoria, que actúa como ordenador central o anfitrión para los dispositivos GPU.
Los dispositivos GPU empleados para realizar las pruebas van desde una tarjeta doméstica cómo la NVIDIA GeForce GTX480, hasta tarjetas de cálculo científico sin salida gráfica y altas prestaciones como las NVIDIA Tesla K20c y K40c. En la tabla 4 se describe el modelo, número de núcleos, memoria global, ancho de banda de memoria y frecuencia de reloj de la GPU.
Los conjuntos de prueba son ejecutados variando el número de moléculas del conjunto de datos y eligiendo diferentes descriptores moleculares como predictores para resolver nuestro problema de clasificación (solubilidad o insolubilidad). Por ejemplo, a partir de descriptores moleculares simples del tipo CPSA (Msurf, Mpola, Msolu, AlogP) o descriptores constitucionales (Natom, MolWe, NRing, NArRg, NRotB, NHDon, NHAcc) que tienen sólo una característica molecular. Por otra parte, se aborda también el cálculo de descriptores basados en huella/fragmemento, con vectores con varias características moleculares, que son computacionalmente intensos. Las combinaciones de descriptores constitucionales (MNBH, MNB, NBH, mon) o combinaciones de descriptores de fragmento/huella (EAE246, EA, AE246) son, a su vez, combinaciones de los descriptores simples anteriores, pero necesitan más tiempo de proceso. En trabajos anteriores, hemos demostrado que la selección de variables es un paso muy importante para una mejor predicción [23].
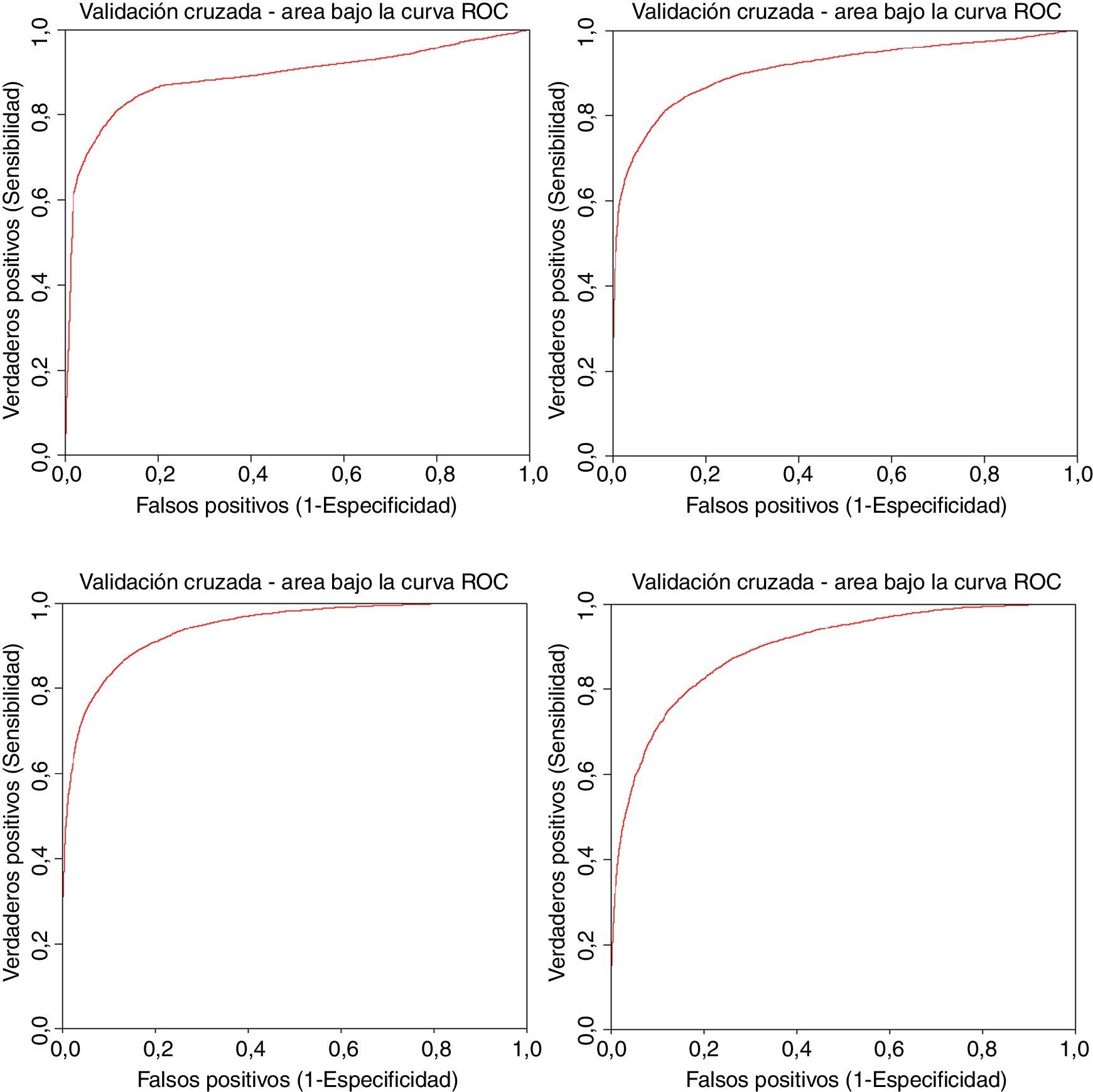
4.1Capacidad predictiva de solubilidad de MSV y GMSVEn esta sección se presentan los resultados que demuestran la capacidad predictiva de las máquinas de soporte vectorial. Para ello se ha empleado la base de datos NCI mencionada anteriormente y se han obtenido curvas ROC que determinan la capacidad predictiva del método para descriptores simples (ALOGP) y complejos (EAE246) en implementaciones secuencial y paralela (fig. 3). En todos los casos se realiza validación cruzada con 5 iteraciones.
 en versión secuencial y paralela (superior izquierda y derecha respectivamente) y para descriptor complejo EAE246 versión secuencias y paralela (inferior izquierda y derecha respectivamente).")
Como puede observarse, en todos los casos se alcanzan valores de área bajo la curva superiores a 0,9 y no hay pérdida de capacidades predictivas debida al proceso de paralelización del algoritmo de máquinas de soporte vectorial. De esta manera demostramos por una parte la bondad de las MSV para predecir la solubilidad contra una base de datos de gran tamaño y empleando tanto descriptores simples como complejos y la conveniencia del empleo de procesadores gráficos para poder reducir el coste temporal de dichas predicciones.
4.2Paralelización y aceleración sobre GPUSe han realizado diferentes pruebas para diferente número de moléculas del conjunto de datos, obteniendo el tiempo de ejecución de MSV con versiones CPU y GPU. Como predictor (datos de entrada), elegimos un descriptor molecular computacionalmente moderado (ALogp, Coeficiente de partición) y un descriptor molecular computacionalmente intenso (EAE246, Contador de estados (EstCt) + AlogP2 Contador de estados (AlCnt) + Huella de conectividad extendida (ECFP)).
Podemos observar que el factor de aumento de velocidad entre la GPU y la CPU aumenta en relación al número de moléculas en el conjunto de datos. Esto es debido a que, al aumentar el número de moléculas utilizadas, también lo hace el número de bloques de hilos que se ejecutan en paralelo, y entonces los recursos de la GPU se utilizan completamente. Sin embargo, para conjuntos de prueba más pequeños, los recursos de la GPU no se utilizan plenamente. Para descriptores molecular sencillos (ALOGP) y un gran número de moléculas, obtenemos unos tiempos de ejecución inferiores para las GPU y aumentos moderados de velocidad de entre 2-4x dependiendo del modelo (GPU vs. CPU, véase figs. 4 y 5). Para los descriptores moleculares complejos, incluso con un pequeño número de moléculas utilizadas, el aumento de velocidad de la versión GPU es muy significativo. Como se muestra en las figuras 6 y 7 para un gran número de moléculas y descriptores complejos (EAE246), tenemos tiempos de ejecución muy inferiores para la GPU y aceleraciones de hasta 45x para los modelos de gama alta (GPU vs CPU).
.")
.")
.")
.")
Sin embargo, cómo puede apreciarse en las gráficas de tiempos de ejecución y aceleración, el uso de procesadores gráficos más potentes y de mucho mayor coste, como K20 y K40, no supone una mejora importante en la ya apreciable aceleración obtenida con la versión paralela sobre una unidad de procesamiento gráfico doméstica como la GTX 480.
5Conclusiones y trabajos futurosEn este trabajo hemos introducido los beneficios del uso de máquinas de soporte vectorial para la optimización de la predicción de la solubilidad de compuestos.
Las MSVs se han entrenado con una base de datos de compuestos solubles e insolubles conocidos, y esta información has sido posteriormente empleada para mejorar la predicción del CV con arquitecturas paralelas. Los resultados obtenidos para las versiones de GPU son prometedores, con unos valores de aceleración obtenidos de hasta 45x, en comparación con la versión secuencial.
Los buenos resultados obtenidos permiten el uso de grandes bases de datos para la predicción de la solubilidad, utilizando métodos de inteligencia computacional. Estos métodos se adaptan bien en la arquitectura GPU para un gran número de moléculas, o descriptores complejos que son necesarios para obtener buenos valores en la predicción.
Como trabajos futuros se pretenden evaluar otros métodos de inteligencia computacional, para la clasificación, regresión o la selección de variables y se emplearan nuevos dispositivos paralelos.
Este trabajo ha sido parcialmente financiado por los proyectos: NILS Mobility Project 012-ABEL-CM-2014A y Fundación Séneca 18946/JLI/13. Los experimentos han sido desarrollados gracias a la generosa donación de hardware de NVIDIA.