


La vasculitis retiniana se caracteriza por inflamación de los vasos de la retina, los síntomas clásicos son disminución de la visión con dolor leve o ausente. También se presenta un escotoma central o paracentral por edema papilar o macular, además de miodesopsias y metamorfopsias. Puede ser una manifestación de enfermedades sistémicas, autoinmunes o infecciosas, o bien se conoce a la vasculitis idiopática.
Se presenta una vasculitis hereditaria que afecta a un niño de 12 años de edad, con el antecedente de ser hijo de una mujer que presentó la misma enfermedad.
Revisando la literatura médica de las vasculitis primarias de origen genético, son muy raras, por lo cual se documenta este caso.
Retinal vasculitis is characterized by inflammation of the retinal vessels. Classic symptoms are diminished eyesight, little or no pain and central or paracentral scotomas due to papillary or macular edema, besides floaters. It can be idiopathic or be seen as a manifestation of autoimmune or infectious systemic diseases. We present the case of a 12 year old boy with hereditary retinal vasculitis, son of a woman with the same disease.
We report what we consider to be a highly unusual case of genetic primary retinal vasculitis.
¿ Introducción
La vasculitis retiniana es definida como una afección inflamatoria de los vasos retinianos. El oftalmólogo diagnostica vasculitis al observar envainamientos vasculares y pocas veces tiene el diagnóstico histopatológico, como puede ser vasculitis leucocitaria, angeítis granulomatosa, arteritis de células gigantes, vasculitis necrosante sistémica o bien, tromboangeítis obliterante. Son muchos los procesos patológicos que pueden afectar la vasculatura retiniana y simular una vasculitis, estrictamente, se excluyen procesos no inflamatorios como la arterioesclerosis, anormalidades congénitas o incremento de la viscosidad sanguínea. La inflamación puede afectar arterias, venas y capilares, aunque es más común la inflamación venosa.
Los síntomas visuales en la vasculitis suelen ser baja de visión, visión borrosa, percepción de luces intensas (fotopsias) y de imágenes móviles cuando la luz es muy blanca (miodesopsias). Algunos pacientes son asintomáticos. Se puede acompañar de alteraciones específicas como los seudoexudados cotonosos, que indican isquemia retiniana, o bien las hemorragias como manchas de Roth, oclusiones venosas, edema macular o edema de la papila óptica. En la vasculitis es frecuente la presencia de leucocitos en el vítreo o en el humor acuoso. Muchas veces se clasifica como idiopática por la falta de alteraciones sistémicas. Otras veces se asocia con enfermedades autoinmunes como el lupus eritematoso, la enfermedad de Behçet, la sarcoidosis y la esclerosis múltiple.1,2
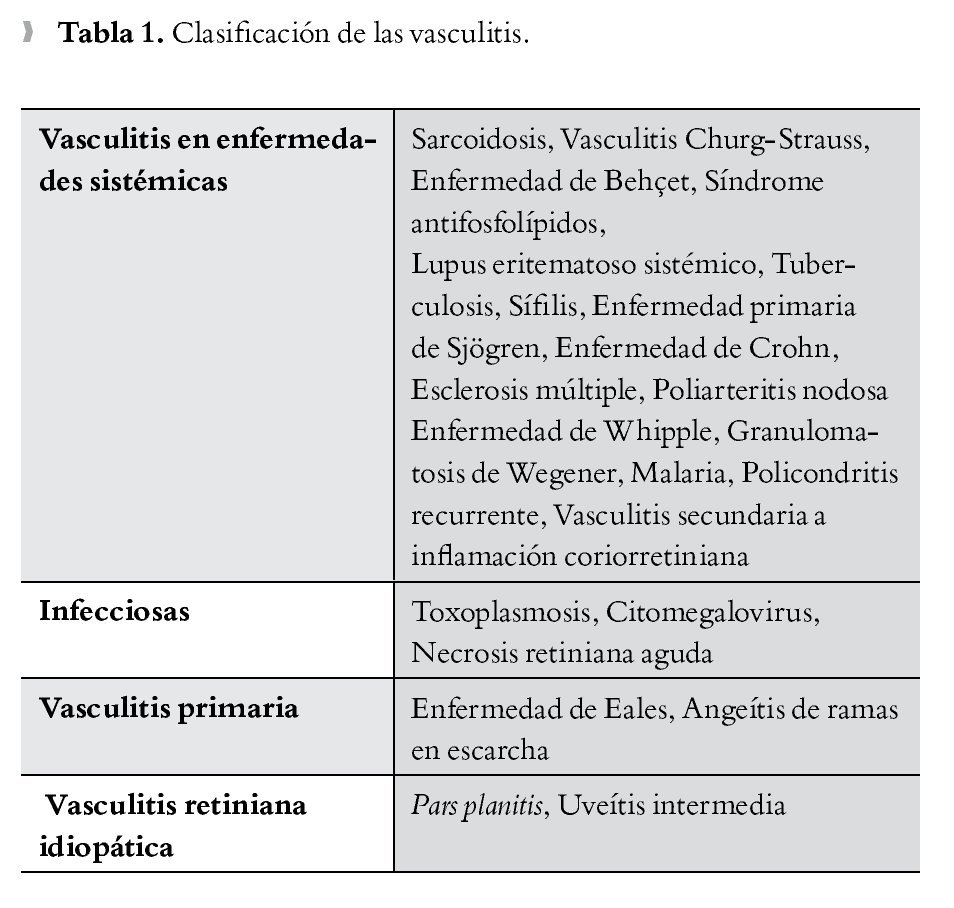
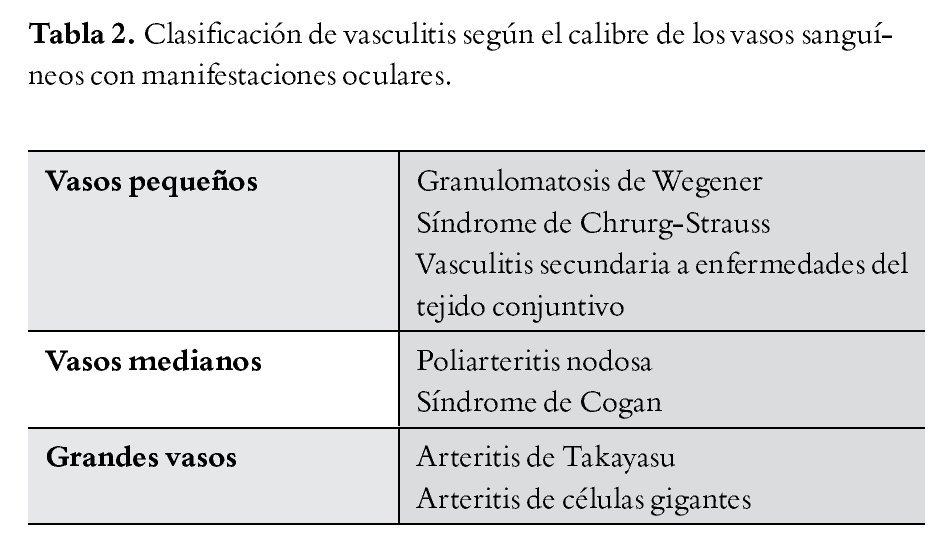
Para entender las vasculitis retinianas, lo más práctico es clasificarlas, una de las más completas es según su etiología (Tabla 1),3 y la más conocida desde hace tiempo es la vasculitis según el grado del calibre vascular (Tabla 2).
Algunos estudios indican que las vasculitis no infecciosas como la pars planitis, y la enfermedad de Eales, son enfermedades autoinmunes inducidas por bacterias que se identifican con algunos de los HLA y polimorfismos de citocinas.1,4-6
Sin datos específicos que ayuden al diagnóstico etiológico, los estudios de laboratorio y gabinete son útiles, por ejemplo una placa simple de tórax con adenopatías sugiere sarcoidosis, las lesiones micronodulares en pulmones sugieren tuberculosis miliar o bien, hay pruebas específicas como la FTA-abs para sífilis y la prueba de ELISA para toxocariasis y toxoplasmosis.
La PCR (reacción en cadena de las polimerasas) del vítreo y el humor acuoso, es la prueba de preferencia en casos persistentes de inflamación intraocular.
¿ Presentación del caso
Paciente del sexo masculino de 12 años de edad, conocido en la Clínica de Uveítis del Hospital General de México "Dr. Eduardo Liceaga" (HGMX) desde marzo del 2010, con el antecedente de notar baja de visión de ambos ojos, con percepción de miodesopsias un año y medio antes de la consulta. No refiere enfermedades asociadas antes o durante su problema ocular, lo importante es que su progenitora tuvo pérdida progresiva de la visión de ambos ojos durante su juventud.
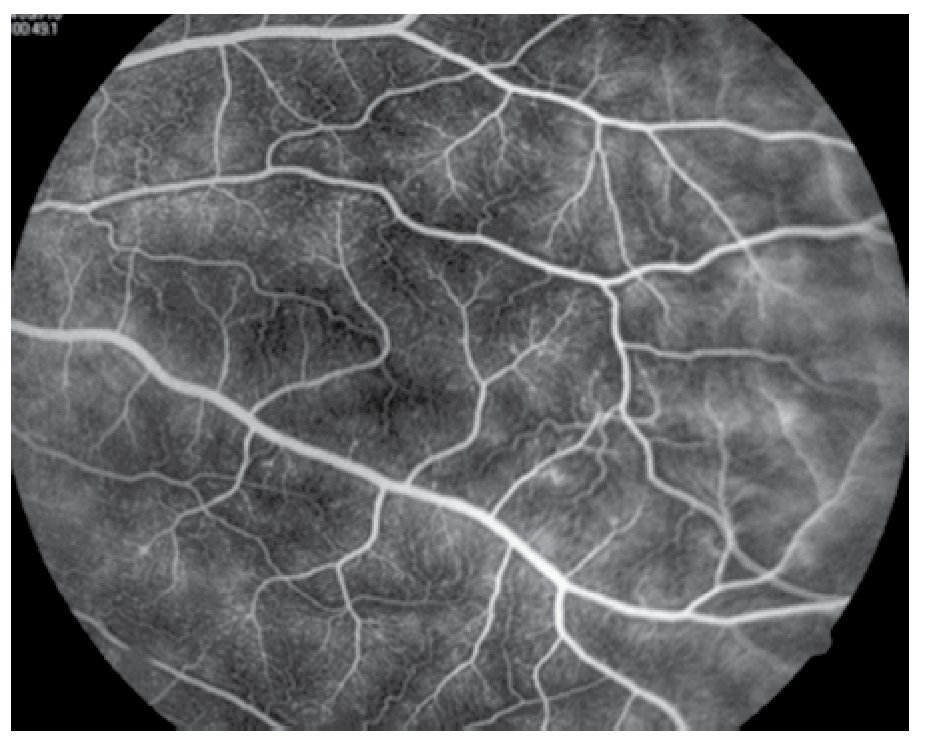
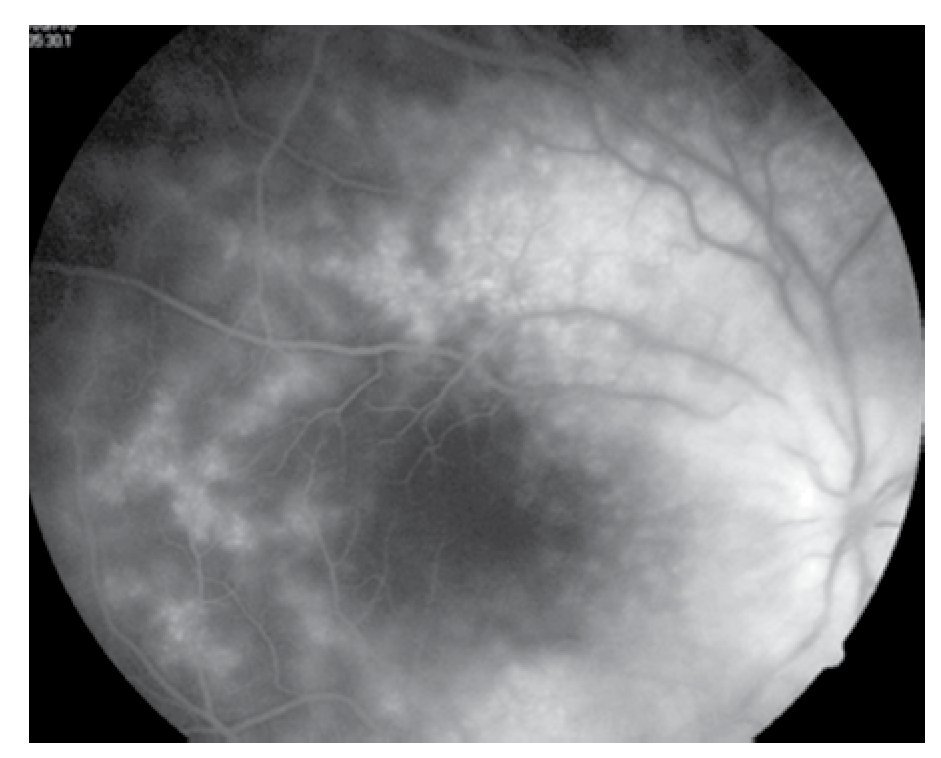
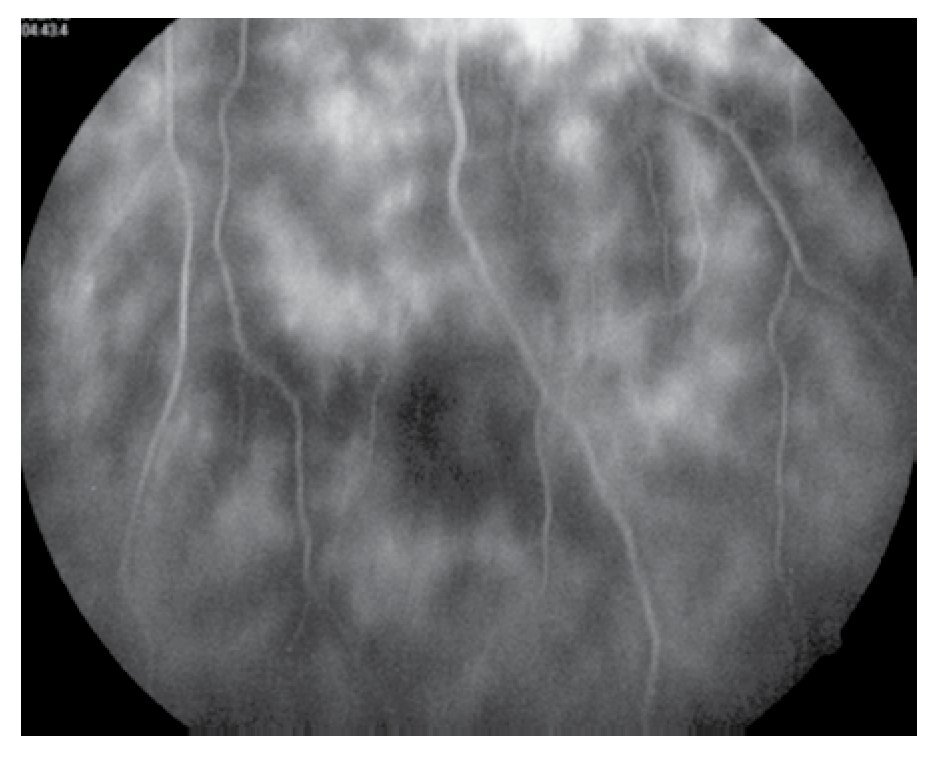
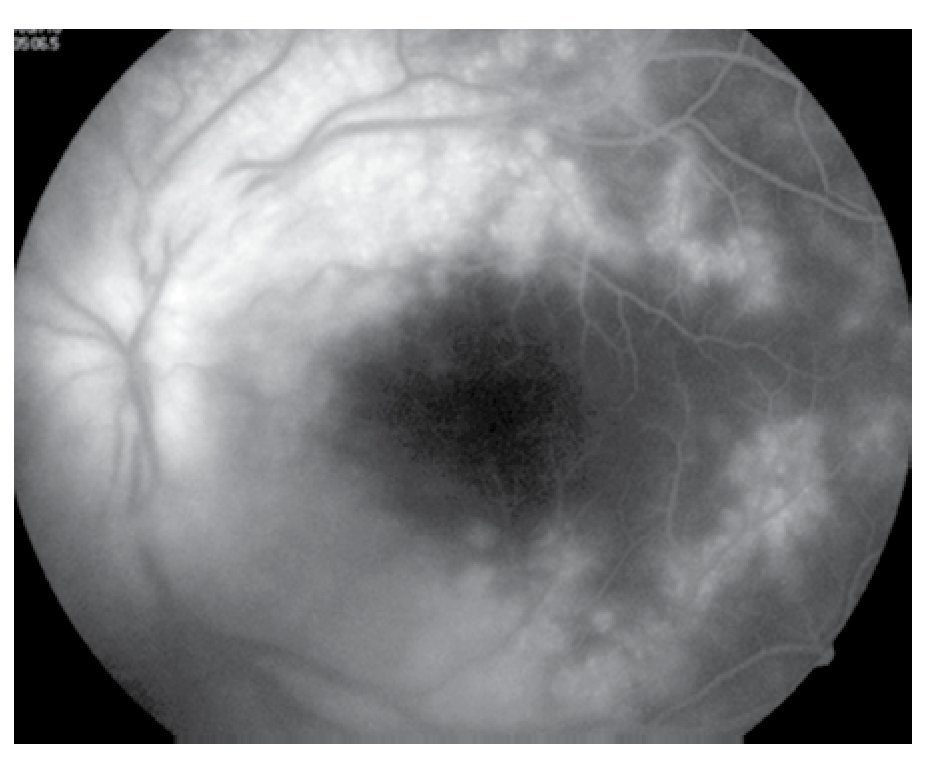
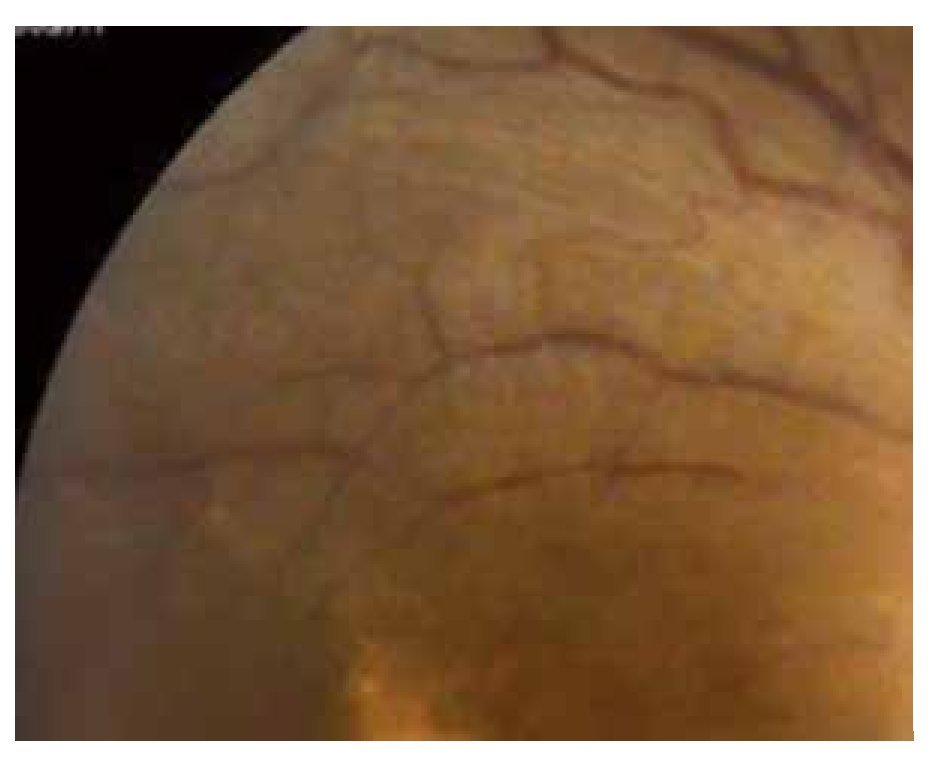
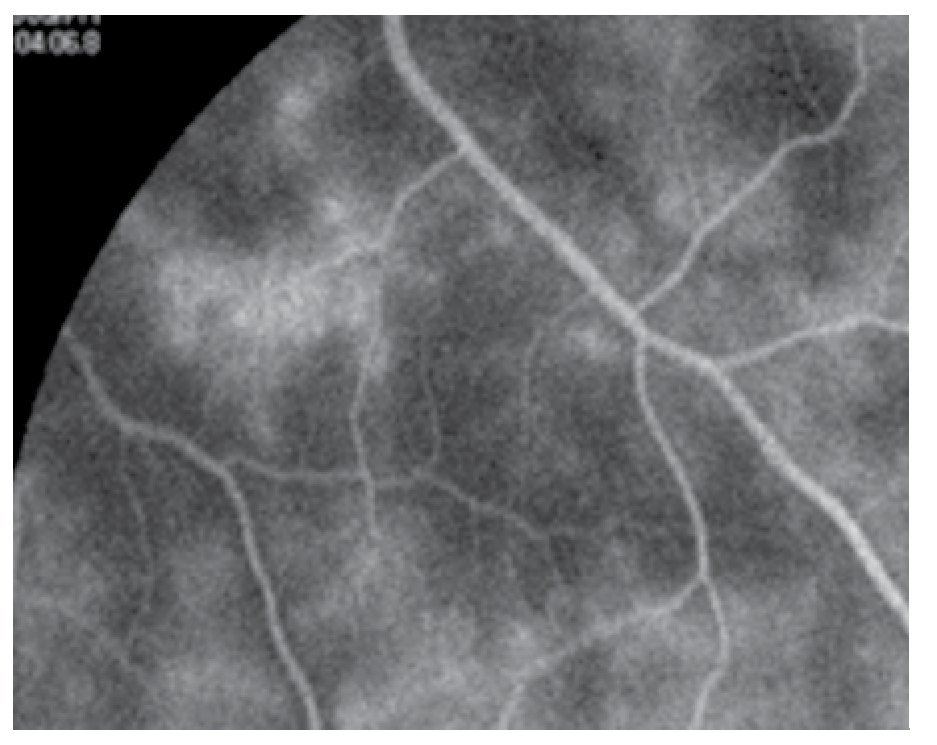
El paciente ya había sido tratado en otro centro hospitalario con prednisona 20 mg y azatioprina 125 mg por día, refiriendo persistente baja de visión de predominio en el ojo derecho (OD), por lo que acude a nuestra institución. En la exploración oftalmológica se encuentra agudeza visual (AV) de 20/140 en el OD y 20/50 en ojo izquierdo (OI), presión intraocular de 12 mmHg en ambos ojos. Segmento anterior normal, sin alteraciones en córnea o en el iris. El vítreo del OD con organización leve y en el OI moderada, sin "copos de nieve" o bien, otro tipo de alteración vítrea. En el fondo de ambos ojos se observa borramiento de los bordes de la papila óptica, retina pálida con imagen de "estrella macular" en el OD y el resto de la retina de los dos ojos con adelgazamiento generalizado de los vasos sanguíneos (Figuras 1 y 2). Las imágenes de la fluorangiografía (FAG) demuestran datos de vasculitis retiniana en toda la retina, con daño de epitelio pigmentado en la mácula, respetando la zona foveal (Figuras 3 a 6).
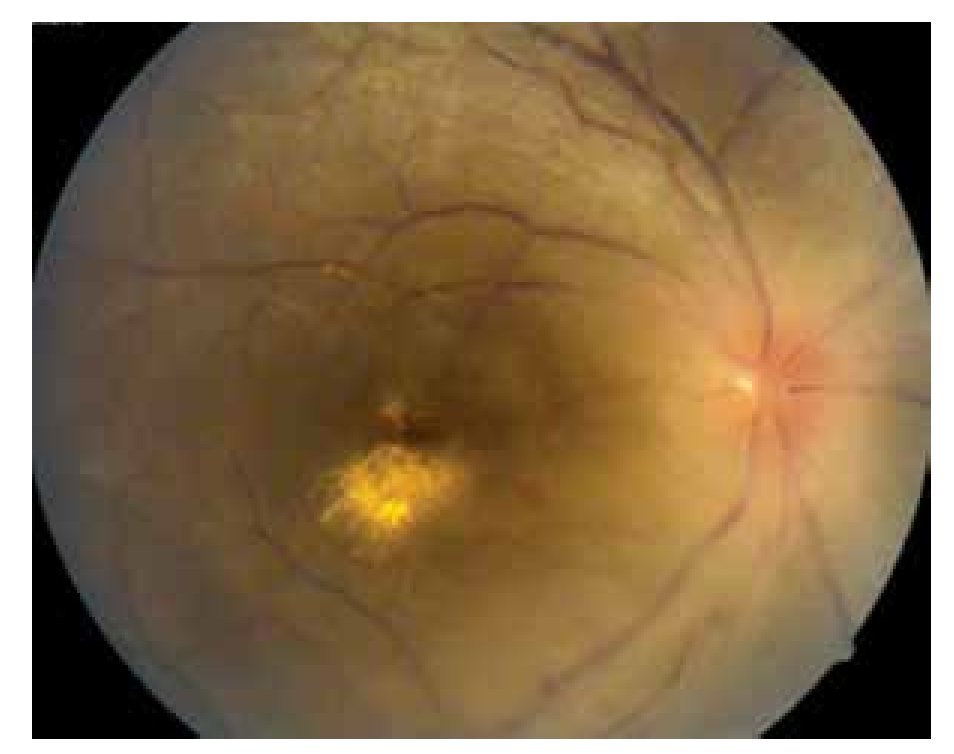
Figura 1. Ojo derecho junio 2010.
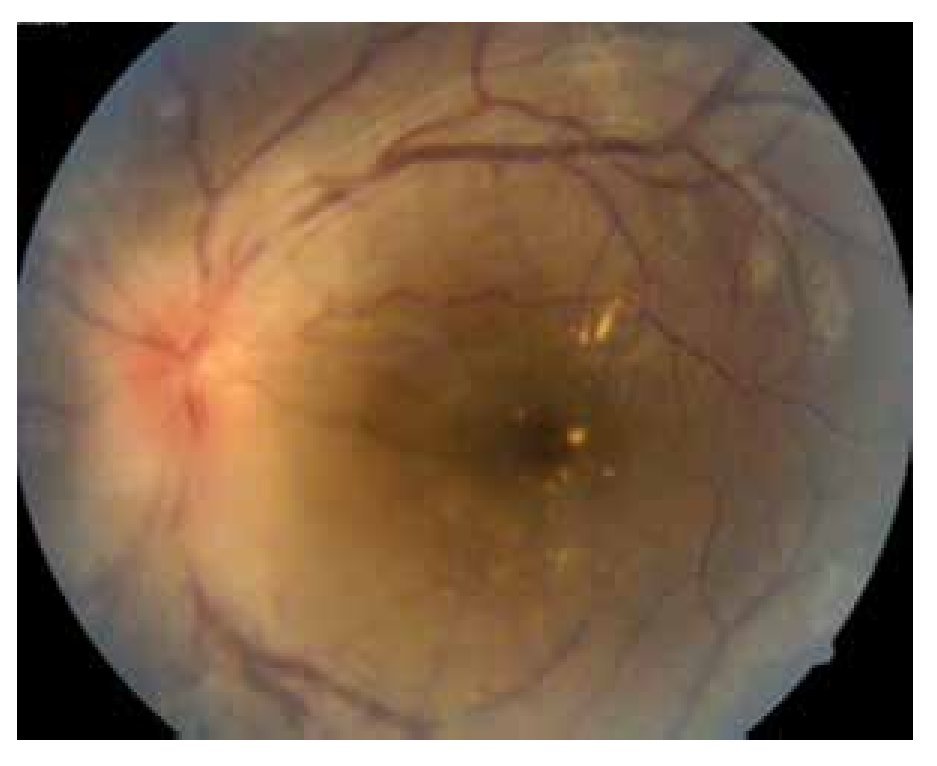
Figura 2. Ojo izquierdo junio 2010.
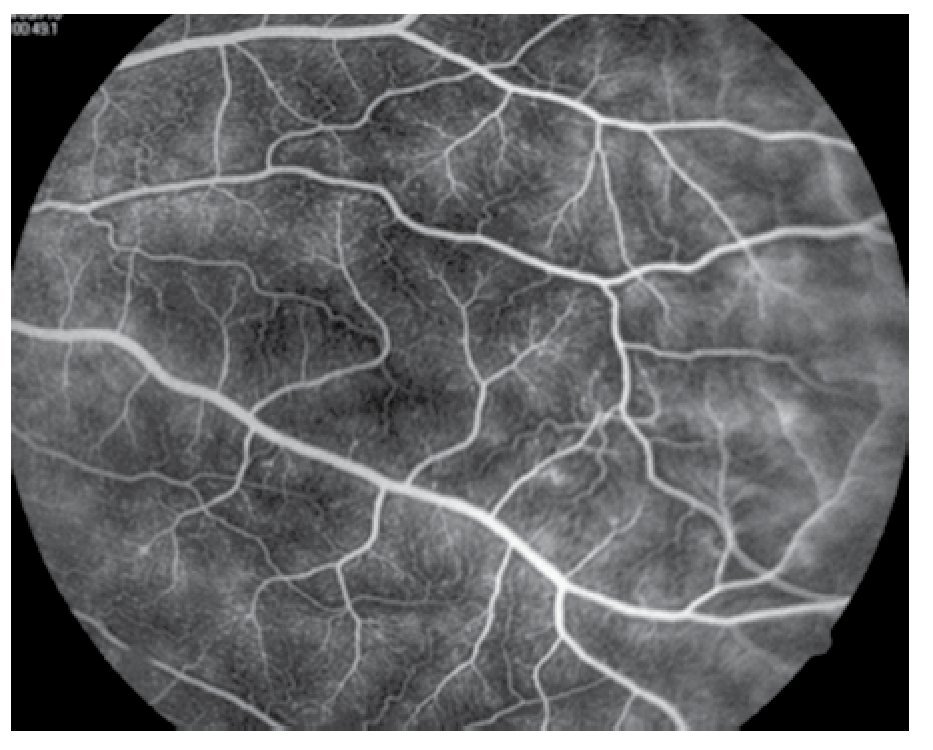
Figura 3. Fluorangiografía del ojo derecho, imagen de vasculitis activa.
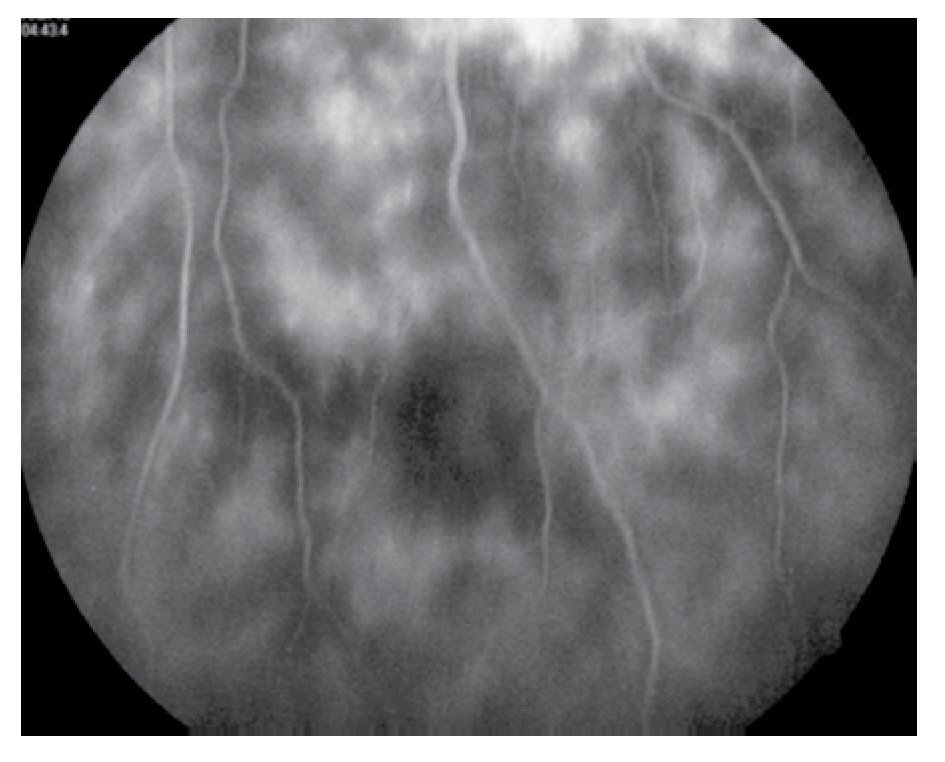
Figura 4. Fluorangiografía del ojo izquierdo, zona inferior de retina en fases tardías.
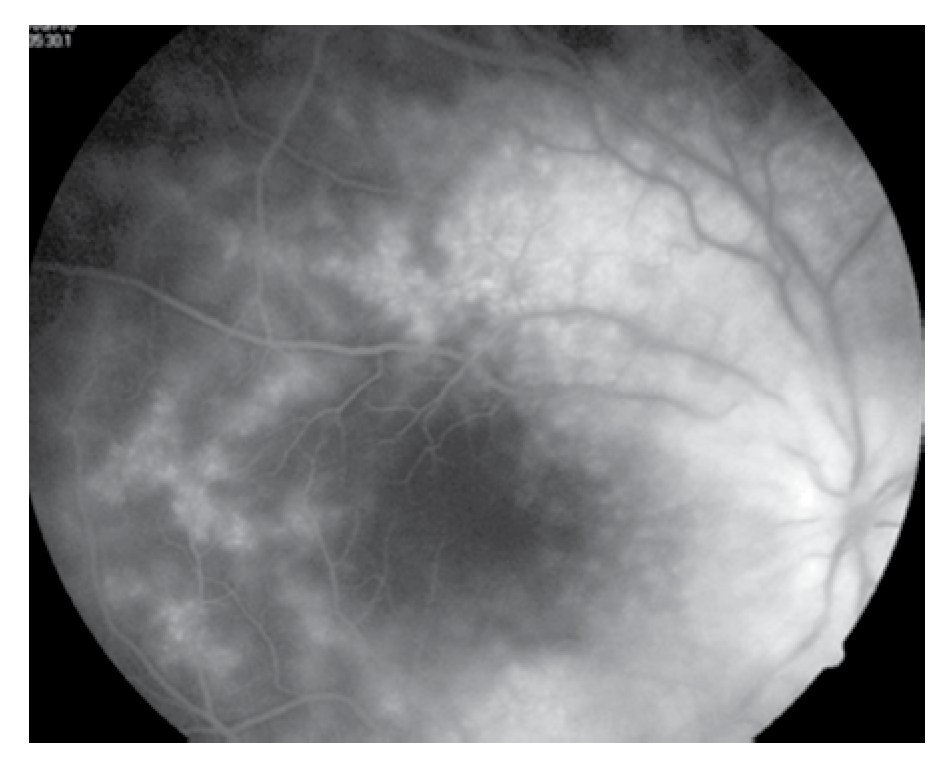
Figura 5. Ojo derecho, mácula en fases tardías.
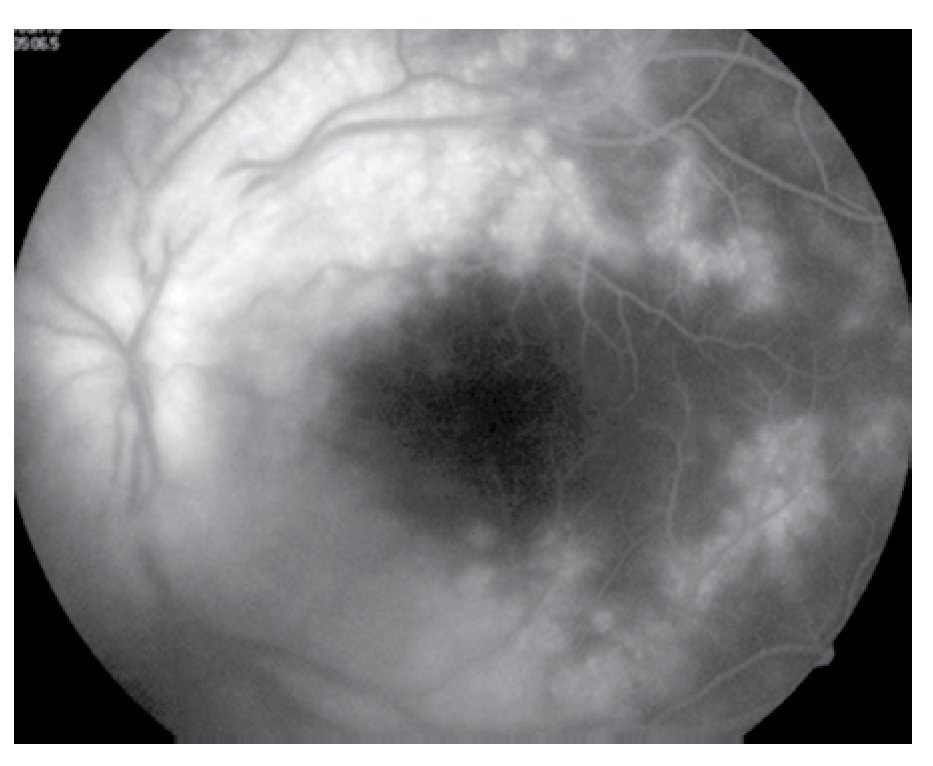
Figura 6. Ojo izquierdo, mácula en fases tardías.
El paciente se envió al Servicio de Pediatría donde le pidieron exámenes de laboratorio (BH, general de orina, PPD, tele de tórax y glucemia) resultando normales, asimismo la exploración física no demostró alteraciones sistémicas.
Con los hallazgos anteriores, se mantiene la terapia inmunosupresora con dosis de reducción durante un año hasta llegar a 5 mg de prednisona y 75 mg de azatioprina por día; mejorando la AV del paciente a 20/60 en ambos ojos.
En el mismo periodo, la madre del paciente es conocida en la Clínica de Uveítis, tiene como antecedente que fue operada del OD de vitrectomía con aplicación de láser a los 29 años de edad, evolucionó pocos años después en percepción y localización de luz en ambos ojos. Solicita cirugía del ojo vitrectomizado por tener una catarata notoriamente cosmética, se hace facoemulsificación sin complicaciones, llevando la AV de percepción de luz a movimiento de manos a un metro; tres meses después es necesario hacer capsulotomía con láser Yag permitiendo valorar el fondo de ojo, el cual se observa de características similares al de su hijo, con papila óptica pálida de bordes borrados, vasos envainados y retina con huellas de fotocoagulación. No es posible fotografiar imágenes por la falta de apertura de la pupila y la capsulotomía.
Se envían a ambos pacientes al Servicio de Genética donde no encuentran relación alguna en su historia familiar, al parecer sólo la madre e hijo (hijo único) son los afectados, no tienen a otro familiar con el mismo problema.
Durante el mes de mayo del 2011, se repite el estudio de FAG notando menos actividad de la vasculitis (Figuras 7 y 8), y en los estudios de fondo de ojo ya no se observan cambios en el vítreo; por lo que se continúa disminuyendo la dosis de la prednisona y la azatioprina hasta mantenerlo sin terapia. Durante un año de seguimiento, el paciente sigue sin cambios en su visión, que es 20/60 en ambos ojos y sin cambios en el fondo de ojo.
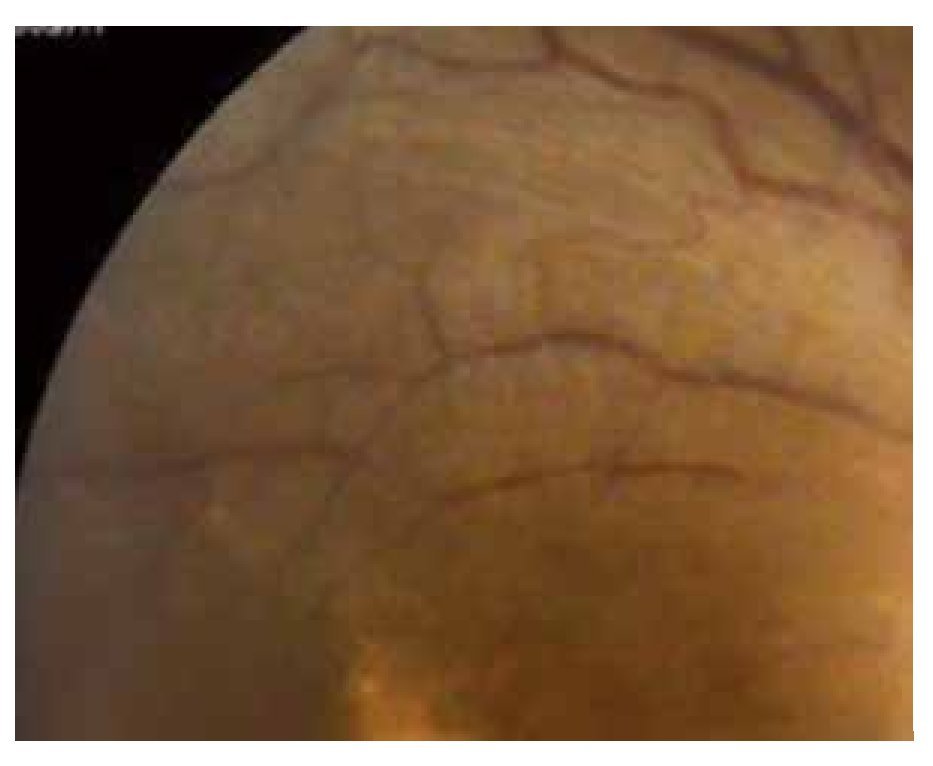
Figura 7. Fondo de ojo derecho, un año después.
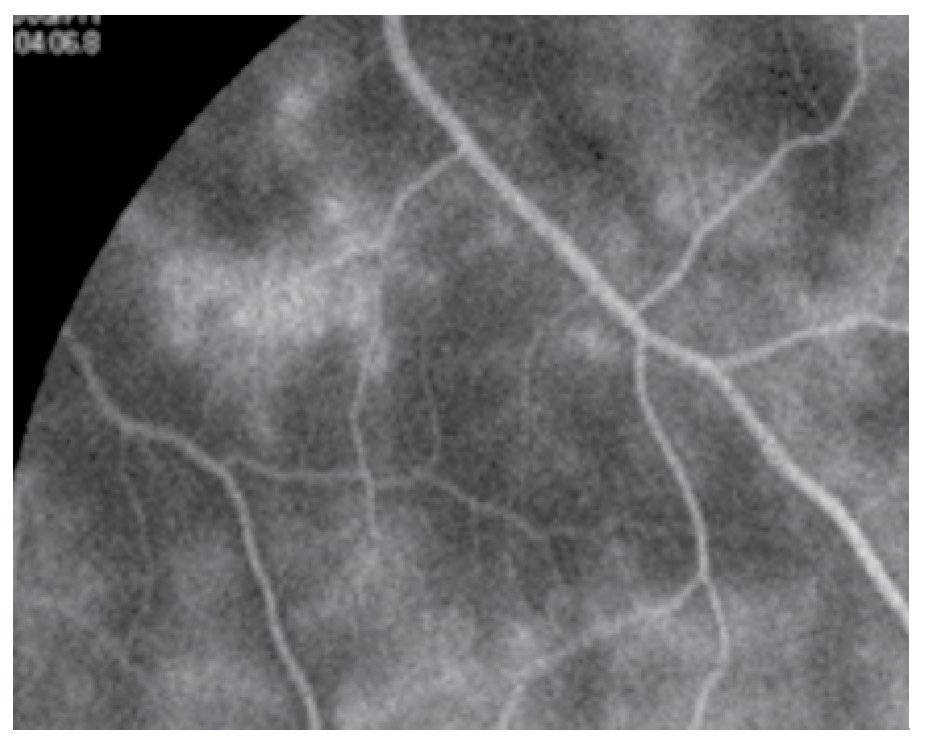
Figura 8. Retina periférica un año después.
¿ Discusión
Las vasculitis primarias son frecuentes en niños y jóvenes, cuando se manifiestan como pars planitis, son comunes las sinequias posteriores, las cataratas y el glaucoma secundario a la terapia esteroidea. Una característica clínica son los "copos de nieve" que corresponden a exudados en vítreo inferior, hay envainamientos venosos con fuga de líquido en fase activa y edema macular cistoide.7
En el caso que presentamos las imágenes de la retina son similares a la pars planitis, con excepción de que el vítreo no presenta alteraciones ("copos de nieve") y que el paciente no ha presentado catarata o hipertensión ocular, incluso nunca presentó reacción inflamatoria de la cámara anterior, hallazgo común de la pars planitis.
Si consideramos las alteraciones retinianas hereditarias, las más comunes son la retinitis pigmentosa, la amaurosis congénita de Leber, la ceguera nocturna congénita estacionaria, la distrofia macular viteliforme y la distrofia macular de Stargardt.8 En el caso que presentamos, una vez descartadas las enfermedades autoinmunes inflamatorias nos quedan las causas hereditarias. La primera es la vitreorretinopatía inflamatoria autosómica dominante. En esta alteración genética los pacientes presentan un marcado edema macular quístico, bilateral, con una profusa exudación de los vasos retinianos, células en el vítreo, en la periferia acúmulos pigmentarios y neovascularización en etapas avanzadas. En el paciente no hay vascularización periférica pero al parecer si la hubo en su mamá, porque le practicaron fotocoagulación retiniana.
Otra posibilidad es un síndrome deinmunodeficiencia variable; ésta es una afección rara, en la cual los pacientes presentan una vasculitis retiniana, bilateral, con un marcado edema macular quístico. El diagnóstico en las primeras fases de la enfermedad es difícil, ya que las únicas alteraciones son tasas bajas de inmunoglobulinas IgG, IgA y/o IgM. Estos pacientes presentan a menudo procesos infecciosos recurrentes, sobre todo pulmonares. A veces se encuentran también alteraciones de los linfocitos B, T reguladores y de los macrófagos.9-11
Otra causa de vasculitis primaria es el síndrome de vasculitis retiniana idiopática con neuroretinitis y aneurismas (IRVAN), este síndrome afecta más a las mujeres, no hay manifestación sistémica y es muy característica la dilatación aneurismática de arteriolas retinianas. En un reporte de un caso de IRVAN, se encontró asociado con anticuerpos antineutrófilos citoplásmicos perinucleares (p-ANCA). En nuestro paciente no observamos dilataciones aneurismáticas, por lo que descartamos se trate de un caso tipo IRVAN.12,13
Un reporte de caso clínico reciente trata sobre una mujer de 35 años de edad con envainamientos muy marcados en la retina inferior del OI, asociado a hemorragias en la misma zona y una hemorragia premacular, no indican tratamiento médico, hacen vitrectomía con recuperación de la visión del ojo afectado; concluyendo se trata de una angeítis de vasos en escarcha.14 Nada parecido al paciente que presentamos.
El tratamiento de la vasculitis retiniana primaria está reservado para los casos con daño en la visión. Los glucocorticoides sistémicos, la azatioprina, el metotrexato, la ciclosporina y la plasmaféresis han sido indicados con eficacia variable. La fotocoagulación retiniana se indica en casos de neovascularización focal, rubeosis iridis y en la hemorragia vítrea. Los casos complicados pueden requerir vitrectomía o cirugía filtrante. En pacientes con edema macular se han aplicado antiangiogénicos como el bevacizumab.15
¿ Conclusión
El paciente presentado en este reporte presenta una vasculitis retiniana primaria tratado con prednisona y azatioprina. De acuerdo a la presencia de alteraciones similares en la retina de su progenitora, aunque no puede considerarse que se trate de una enfermedad mendeliana, existe la posibilidad de una carga genética que este influyendo en la presentación de este caso.
¿ Conflicto de intereses
La autora declara no tener conflicto de intereses.
¿ Financiamiento
La autora no recibió ningún patrocinio para llevar a cabo este artículo.
Correspondencia:
Zempoala 537, Colonia Vértiz Narvarte,
C.P. 03020, México D.F., México.
Correo electrónico: mgtenorio9@yahoo.com