


Las malformaciones oculares congénitas constituyen un amplio grupo de alteracio nes de la organogénesis del ojo, que pueden originarse por la acción de agentes genéticos y/o ambientales durante el desarrollo embrionario. Estas anomalías ocupan uno de los primeros lugares como causa de discapacidad visual o ceguera en niños, contribuyendo de manera significativa a la morbilidad infantil. En la última década se ha generado un conocimiento enorme, acerca de las causas genéticas de diversas alteraciones del desarrollo ocular en el humano. Este conocimiento no sólo ha permitido una mejor comprensión de los procesos embriológicos que llevan al desarrollo de estructuras visuales funcionales, sino que ha impactado de manera directa en el diagnóstico, evaluación y asesoramiento genético de los afectados y sus familias.
Congenital eye malformations are a heterogeneous group of diseases in which embryological eye development is disturbed by genetic, environmental (or a combination of both) factors. These anomalies are among the commonest causes of childhood visual impairment in most populations around the world. In the last few years, the knowledge of the genetic basis of congenital eye malformations has greatly improved by the recognition of mutations in several monogenic forms of these defects. These advances have enhanced not only the understanding of the embryological basis of human eye development but also the clinical approach to this group of patients and their families.
Introducción
Desarrollo ocular normal
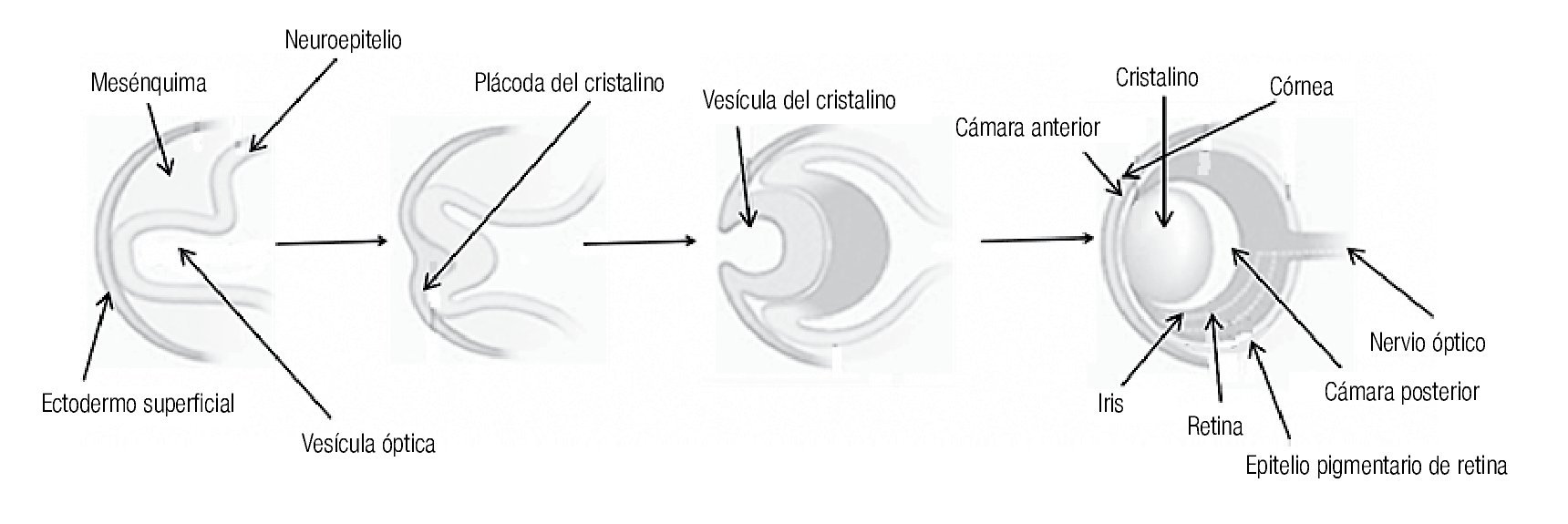
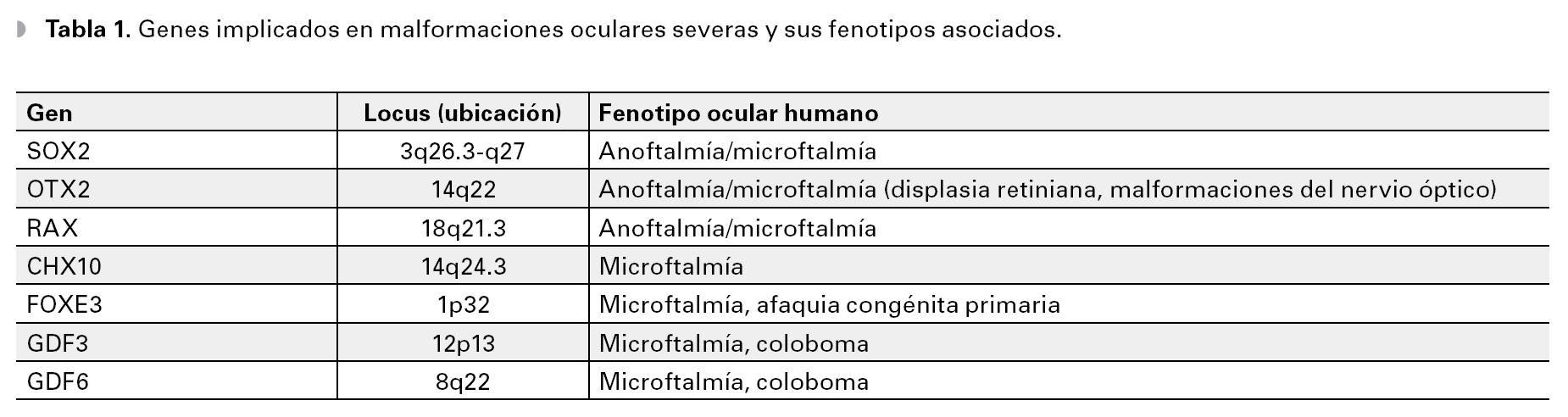
La primera manifestación del desarrollo ocular en el humano, ocurre con la aparición de un surco óptico a cada lado del cerebro anterior (diencéfalo), en el día 26 después de la fertilización. Para el día 28 (semana seis de embarazo), los surcos se transforman en evaginaciones localizadas a ambos lados del cerebro, denominadas "vesículas ópticas". Las vesículas ópticas crecen hacia fuera y hacen contacto con el ectodermo de superficie de la cara del embrión, lo que induce un engrosamiento superficial llamado "placoda cristaliniana", que posteriormente se invaginará para dar origen a la depresión del cristalino (Figura 1). En el día 32, la vesícula óptica se invagina y forma la copa o cúpula óptica de pared doble, que más tarde dará origen a la retina neural (derivada de la pared interna) y al epitelio pigmentario (derivada de la pared externa). En el día 33, la depresión de cristalino, que ha seguido invaginándose, se despega del ectodermo de superficie y se transforma en la vesícula del cristalino (Figura 1). En esta etapa, la copa óptica presenta una hendidura a lo largo de su parte inferior conocida como fisura coroidea.
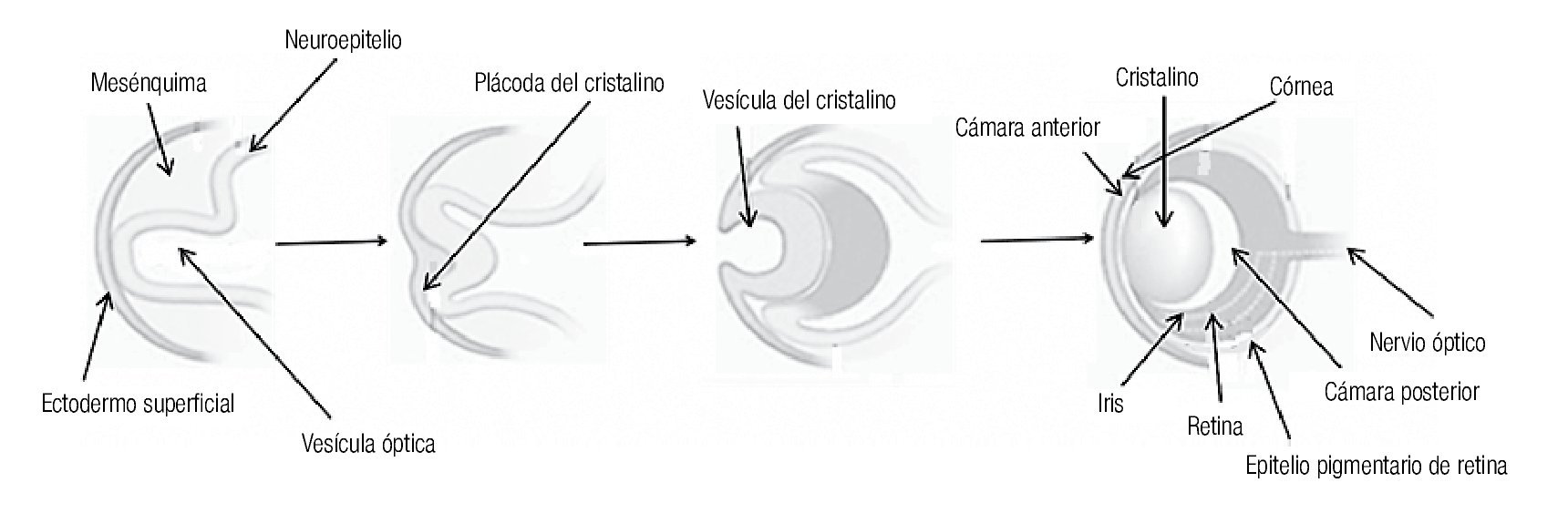
Figura 1. Desarrollo del ojo humano.
En la séptima semana los labios de la fisura coroidea se fusionan, dejando un orificio que formará la pupila. La falla del cierre de la fisura originará un coloboma que puede involucrar al iris, la retina y al nervio óptico. La alteración de cualquiera de los procesos implicados en el desarrollo temprano del ojo, puede llevar a malformaciones severas como anoftalmía o microftalmía. Aunque en la mayoría de los casos, la causa de estas malformaciones es desconocida, se han reconocido diversos factores ambientales y genéticos. Esta revisión está enfocada a los factores genéticos, identificados como causantes de malformaciones oculares en el humano.
La etiología de las malformaciones oculares es compleja e incluye factores ambientales y genéticos. Las causas ambientales pueden corresponder a factores potencialmente teratógenos como agentes químicos, biológicos o físicos, que interrumpen el desarrollo ocular normal.1 Dentro de los factores genéticos pueden reconocerse tres tipos de origen: multifactorial, cromosómico y monogénico. Se considera que la etiología más frecuente de las malformaciones oculares congénitas es la de tipo multifactorial, en la que una combinación de factores genéticos (diversas variantes en un número no precisado de genes, origina una predisposición) y factores ambientales, originan la malformación. Diversas anomalías cromosómicas (cromosomopatías) han sido demostradas en pacientes con malformaciones oculares severas, sugiriendo la presencia de genes relacionados a organogénesis ocular en los cromosomas o regiones cromosómicas implicadas.2,3 En las alteraciones oculares de origen monogénico, la mutación de un solo gen ocasiona la anomalía. Las alteraciones multifactoriales y las cromosómicas por lo general, tienen un riesgo bajo de heredarse a la descendencia. Sin embargo, las causas monogénicas pueden tener riesgos muy elevados de repetirse en los hijos de los afectados. Por esta razón, la identificación de la etiología de la malformación ocular es de gran trascendencia para el manejo de los pacientes y sus familias.4
Esta revisión se enfoca a las causas monogénicas de malformaciones oculares severas, que incluyen la ausencia de estructuras oculares (anoftalmía), la presencia de ojos poco desarrollados y con malformaciones acompañantes (microftalmía), y la presencia de fisuras oculares resultado de una falla en el cierre de la fisura coroidea (coloboma).
Anoftalmía, microftalmía y coloboma
Las malformaciones oculares pueden ocurrir de manera aislada o como parte de síndromes genéticos complejos.5 Entre las anomalías congénitas oculares se incluyen la microftalmía, la anoftalmía y el coloboma, por mencionar algunas.
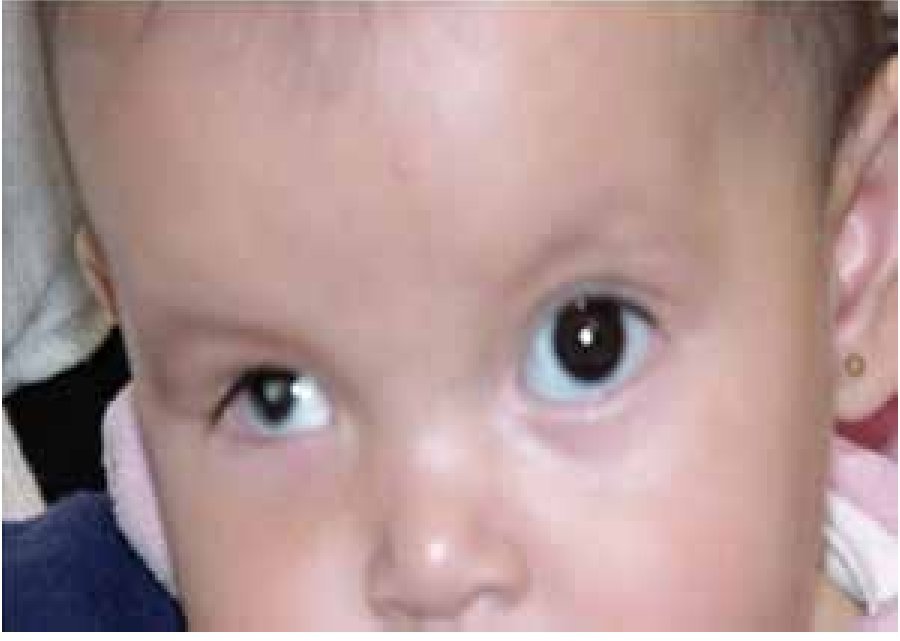
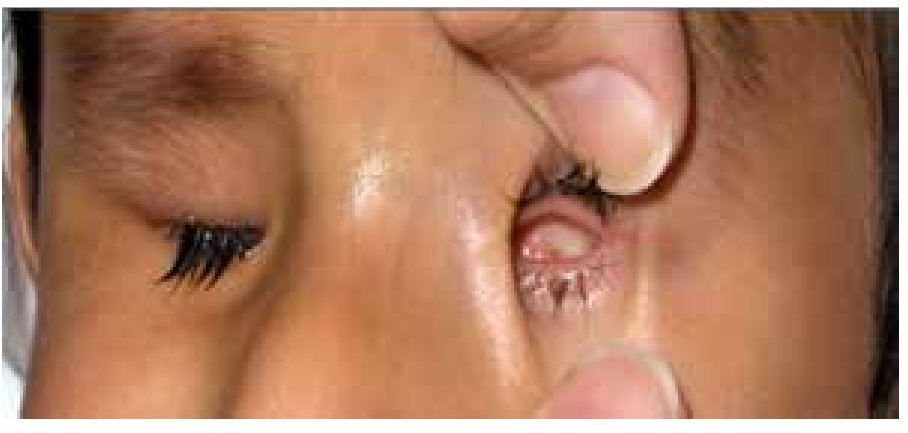
La microftalmía (Figura 2) se define como un ojo, con una longitud axial dos desviaciones estándar por debajo del promedio para la edad.3 Generalmente, el lado afectado de la cara está infradesarrollado y la órbita es pequeña. La microftalmía se puede asociar a otras anomalías congénitas oculares y extraoculares. Algunos casos de microftalmía pueden estar asociados con un quiste, que se cree sea el resultado de un fracaso en el cierre de la fisura óptica.6,7 La microftalmía grave se debe a la interrupción del desarrollo del ojo. La malformación ocular más severa es la anoftalmía (Figura 3), definida por la ausencia congénita de globo ocular con presencia de anexos oculares (párpados, conductos lagrimales).5 En algunos casos se puede reconocer el tejido ocular, solamente desde el punto de vista histológico. En la anoftalmía primaria, el desarrollo del ojo se interrumpe al comienzo de la cuarta semana y es debida a una falla en la formación de la vesícula cristalina, mientras que en la anoftalmía secundaria se suprime el desarrollo del prosencéfalo, y la ausencia de uno o ambos ojos es una de las distintas anomalías asociadas.8
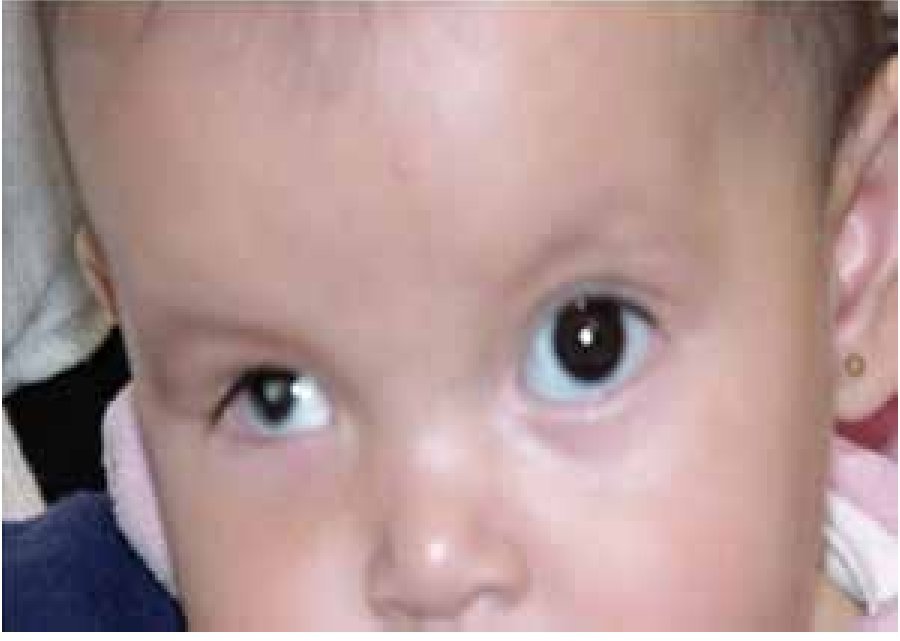
Figura 2. Paciente con microftalmía de ojo derecho.
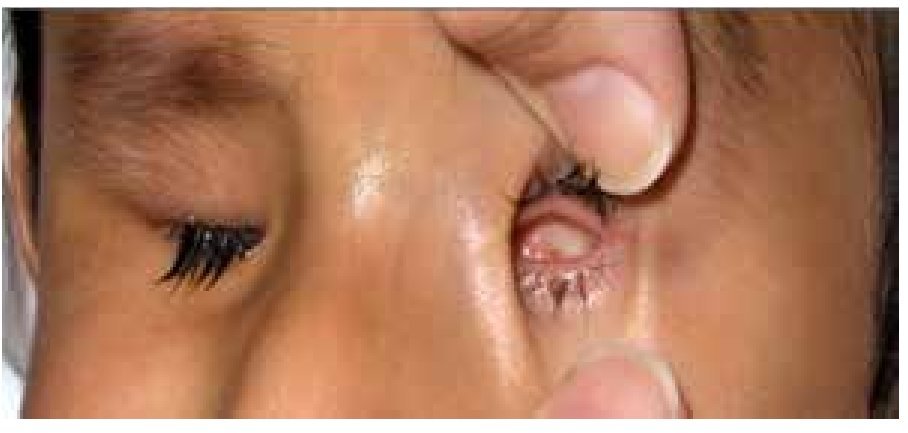
Figura 3. Anoftalmía unilateral izquierda.
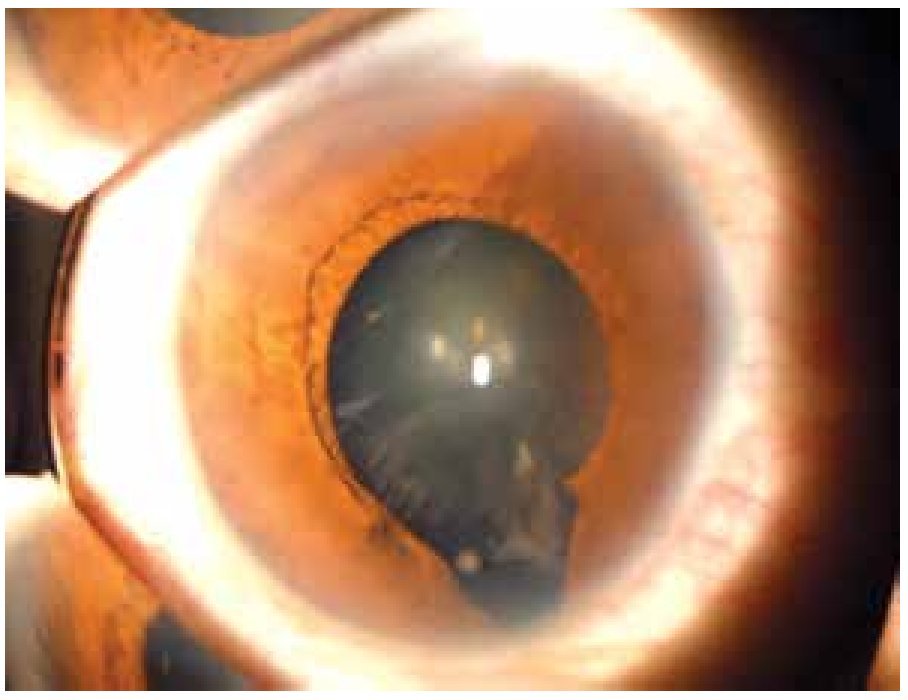
El coloboma (Figura 4) es una anomalía del sector inferior del iris o bien, una escotadura en el margen pupilar que otorga a la pupila un aspecto de cerradura. Un coloboma típico se debe a la falta de cierre de la fisura coroidea durante la sexta semana de desarrollo, y puede involucrar diversas partes del ojo, el iris, la retina, el nervio óptico y el cuerpo ciliar.9,10 Los defectos clásicos son la ausencia parcial del cuadrante nasal inferior del iris, la coroides y la retina, y se asocian frecuentemente a microftalmía, anoftalmía y/o cataratas.11 El quiste colobomatoso es un subtipo de coloboma ocular, en el cual se observa un quiste retrobulbar en combinación con defectos del iris y de las coroides. El coloboma ocular puede manifestarse solo (aislado) o bien, ser un signo de síndromes como CHARGE (coloboma, enfermedades del corazón, atresia de coanas, retraso del crecimiento, retraso y/o anomalías del sistema nervioso central, hipoplasia genital y anomalías del oído y/o sordera).12 El coloboma ocular puede presentarse de forma esporádica o bien, ser transmitido de manera autosómico recesiva, autosómico dominante o ser ligada al cromosoma X.9
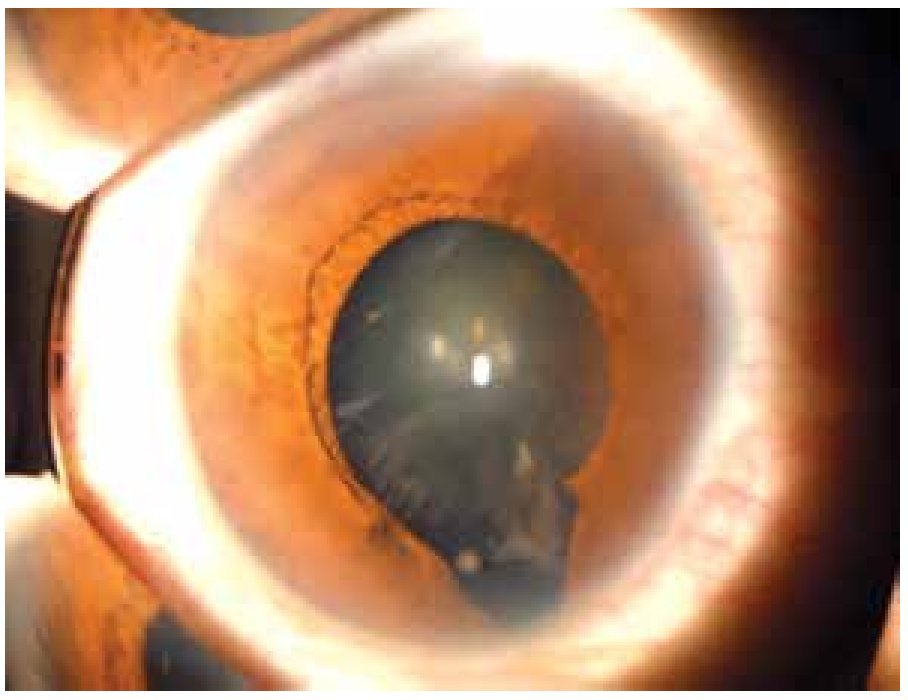
Figura 4. Coloboma de iris y cristalino (cortesía de la Dra. Rocío Arce).
Genes asociados a formas monogénicas de malformaciones congénitas
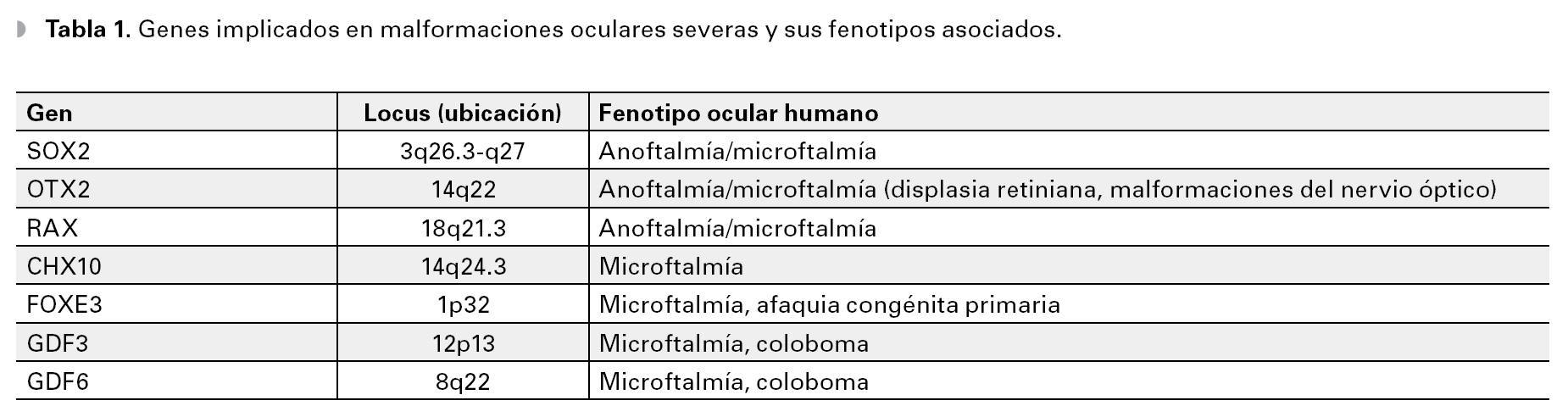
En los últimos años se han reconocido diversos genes (Tabla 1) responsables, de diversos defectos en el desarrollo del ojo en el humano. Se ha determinado que mutaciones en estos genes, explican del 18% al 25% de los casos de malformaciones oculares severas.13
SOX2
SOX2, localizado en el cromosoma 3q26, es el gen más importante de este grupo, ya que es responsable de aproximadamente 10% a 20% de casos de anoftalmía y microftalmía bilateral.14,15SOX2 codifica un factor de transcripción con una función primordial en el desarrollo embrionario en numerosos tejidos, incluyendo el ojo. SOX2 trabaja de manera cooperativa con PAX6, para regular otros genes que promueven el desarrollo del cristalino.16 Se han descrito diversas mutaciones dominantes de este gen en sujetos con anoftalmía o microftalmía, incluyendo deleciones completas del gen, mutaciones puntuales y deleciones parciales.14,17-20 Una deleción de 20 bases en el extremo 5' del gen, es una de las mutaciones más frecuentemente identificadas.14,15,18,19 Algunos sujetos con mutación en SOX2, presentan el llamado "síndrome por deficiencia de SOX2", que además de malformaciones oculares incluye retraso mental, anomalías neurológicas, dismorfias faciales, retraso del crecimiento posnatal, anomalías esofágicas y criptorquidia.21,22
OTX2
El gen OTX2, localizado en 14q22, codifica un factor de transcripción necesario para el desarrollo embrionario de las estructuras cefálicas en vertebrados.23 Se han descrito diversas mutaciones dominantes de OTX2, en pacientes con anoftalmía o microftalmía asociada a malformaciones del sistema nervioso central y retraso mental.24-26 También se han descrito casos con displasia retiniana.27 Se ha determinado que aproximadamente 3% de sujetos con anoftalmía/microftalmía bilateral, tienen alteración en OTX2.14
RAX
RAX, localizado en el cromosoma 18q21.32, es otro gen esencial para el desarrollo ocular, posiblemente a través de su función en el establecimiento y la proliferación de células progenitoras de la retina.28 Aproximadamente, 2% de sujetos con anoftalmía y microftalmía presentan mutaciones en RAX.29-31 A diferencia de los genes mencionados anteriormente, las mutaciones en el gen RAX siguen un patrón de herencia autosómico recesivo.
CHX10
Este gen, también conocido como VSX2, se localiza en 14q24 y codifica una proteína que permite la proliferación de células precursoras neurorretinianas.32 El fenotipo ocular asociado a mutaciones en CHX10 es heterogéneo, ya que se han descrito casos de microftalmía, coloboma y catarata.33-35 Aproximadamente 2% de sujetos con microftalmía presentan mutaciones recesivas en CHX10.33 Sin embargo, en un estudio reciente en 50 sujetos mexicanos con anoftalmía/microftalmía, no se identificó ninguna mutación en este gen.31
FOXE3
El gen FOXE3 codifica un factor de transcripción específico del desarrollo ocular, altamente conservado en la filogenia. Mutaciones en FOXE3 originan un espectro amplio de malformaciones oculares que incluyen anomalías del segmento anterior, afaquia congénita, esclerocórnea, cataratas y microftalmía.36-40 En un estudio reciente, aproximadamente 25% de sujetos con microftalmía presentaron mutaciones recesivas en FOXE3.40 Un aspecto interesante de este gen, es que sus mutaciones heterocigotas (dominantes) también se han asociado a desarrollo ocular anormal, como colobomas coriorretinianos y de iris, anomalía de Peters y cataratas de tipo cerúleo, nucleares y corticales de aparición temprana.38
GDF3
GDF3 es un miembro de la familia de proteínas morfogenéticas de hueso (BMP), las cuales tienen participación en una amplia variedad de procesos durante el desarrollo embrionario.41 El gen GDF3 está localizado en 12p13.1. En 2010, se reconoció que GDF3 está implicado en el desarrollo ocular humano. En un grupo de pacientes con malformaciones oculares, se identificaron cuatro casos con mutaciones dominantes en GDF3, y un espectro amplio de malformaciones oculares, que fueron desde coloboma de iris hasta microftalmía bilateral.42 De manera interesante, se encontró evidencia de falta de penetrancia de algunas de estas mutaciones.42 Los resultados publicados por estos autores, indicaron que GDF3 puede estar mutado en aproximadamente 2% de pacientes con el espectro de microftalmía, anoftalmía y coloboma, y que debe incluirse como uno de los genes a analizarse en sujetos con malformaciones oculares congénitas.
GDF6
GDF6, un gen localizado en 8q22 y altamente relacionado a GDF3, es responsable de hasta 8% de casos de malformaciones oculares congénitas, particularmente microftalmía y coloboma.31,43 Además de las malformaciones oculares, algunos pacientes con mutaciones dominantes en este gen presentan alteraciones esqueléticas y polidactilia, lo que indica la importancia de GDF6, miembro de la familia de proteínas morfogenéticas de hueso, en diversos aspectos del desarrollo embrionario temprano.44
Conclusiones
La realización de estudios genéticos en casos aislados, familias y grupos numerosos de pacientes con malformaciones oculares congénitas, ha permitido el reconocimiento de diversos genes que son esenciales para el desarrollo normal de los ojos, y cuyas mutaciones originan defectos en la organogénesis ocular. Al mismo tiempo, la disponibilidad de los estudios genéticos en un número creciente de centros hospitalarios ha permitido establecer un estimado inicial acerca de la tasa de mutación de cada gen específico, y así realizar una mejor planeación del estudio genético en casos nuevos, dándole prioridad a los genes en los cuales se ha reportado una mayor frecuencia de mutación en diferentes poblaciones, Aunque sin duda, existen muchos más genes por identificarse, el conocimiento alcanzado en los últimos años está permitiendo un mejor abordaje del paciente con malformaciones congénitas oculares. Dados los avances recientes en el reconocimiento de las bases genéticas de las malformaciones de ojo, la recomendación actual es realizar estudio molecular de todo sujeto con anoftalmía/microftalmía unilateral o bilateral. Esta información es de gran importancia no sólo para una mejor comprensión de estas malformaciones, sino para un adecuado asesoramiento genético a las familias afectadas.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Financiamiento
Los autores no recibieron patrocinio para llevar a cabo este artículo.
Correspondencia:
Dr. Juan Carlos Zenteno.
Unidad de Investigación, Instituto de Oftalmología "Conde de valenciana", Chimalpopoca N° 14, Colonia Obrera, C.P. 06800, México. D.F., México.
Teléfono: (55) 5442 1700, ext. 3212.
Correo electrónico: jczenteno@institutodeoftalmología.org
