





La enfermedad de Behçet es una dolencia crónica-recurrente que se caracteriza por la presencia de vasculitis multisistémica con manifestaciones mucocutáneas y oculares. La mayoría de estas se autolimitan sin dejar secuelas, excepto por las oculares. Las variadas manifestaciones clínicas y la falta de pruebas patognomónicas hacen de la enfermedad de Behçet un reto diagnóstico cuya prevalencia en México se desconoce. El propósito de este reporte es la presentación de 2 casos de la enfermedad con diferentes características, por la importancia del diagnóstico, sus secuelas, y además, su baja prevalencia. Se presenta el caso de paciente femenino que ingresa al servicio de oftalmología por hiperemia conjuntival y disminución de agudeza visual de ojo izquierdo de 5 meses de evolución. A la exploración se observan úlceras en mucosa oral, lesiones induradas en miembros inferiores y uveítis. Se realizan pruebas diagnósticas complementarias y se inicia tratamiento, mostrando buena evolución general. A pesar del manejo la mejoría visual es poca. El segundo paciente se presenta en el servicio de urgencias de oftalmología por dolor ocular e hiperemia conjuntival de ojo izquierdo de una semana de evolución. Ya con diagnóstico de enfermedad de Behçet, el paciente presenta úlceras bucales, placas eritematosas en miembros inferiores y uveítis. Se inicia tratamiento tópico con mejoría sintomatológica. Las implicaciones oculares en la enfermedad de Behçet son serias. La inflamación ocular recurrente es común y lleva a un daño permanente a menos que se instituya un tratamiento efectivo. El diagnóstico y tratamiento precoz disminuyen la afección ocular.
Behçet's disease is a cronic-recurrent pathology that is characterized by the presence of multisistemic vasculitis with mucocutaneus and ocular manifestations. Clinical data is mostly selflimited and leaves no permanent damage, except for ocular manifestations. The variety of clinical characteristics and the absence of pathognomonic evidence make Behçet's disease a challenge with unknown prevalence in Mexico. The purpose of this report is to describe two clinical cases that presented with different characteristics, for the importance of correct diagnosis, the knowledgement of its effects, and the low prevalence of the disease. We present a female patient with 5 months of left eye conjunctival redness and decreased visual acuity. At the physical examination, she presents aphthous ulcers in oral mucous, erythema nodosum in legs, and uveitis. Diagnostic tests are made and systemic and local treatment is started with good general results. In spite of the treatment, visual acuity didn’t improve much. Second patient presents in Ophthalmology Urgency Service due to 1 week left eye pain and conjunctival redness. Already diagnosed with Behçet's disease, he presented erythema nodosum in legs, aphthous ulcers and uveitis. Topical treatment is started with clinical improvement. Ocular affection in Behçet's disease is serious. Recurrent ocular inflammation is common and leads to permanent damage unless effective treatment is established. Early diagnosis and treatment of the disease can reduce ocular damage.
La enfermedad de Behçet es una vasculitis sistémica crónica recurrente que se caracteriza por la presencia de úlceras orales y genitales, uveítis y eritema nudoso1. La edad de presentación es entre la segunda y cuarta década de la vida con una presentación familiar del 8-18%2. Aunque la etiología es aún desconocida, se cree que la enfermedad de Behçet es activada por factores ambientales como agentes microbianos en individuos con una genética particular3. Los pacientes con enfermedad de Behçet tienen niveles más elevados de S. sanguis en la flora oral que los pacientes sanos o pacientes con otras enfermedades. También se pueden detectar por PCR virus de herpes simple tipo 1 en saliva, úlceras intestinales y úlceras genitales en pacientes con la enfermedad en comparación con pacientes sanos2.
La enfermedad es más prevalente en el Medio Oriente y Japón, donde hay una asociación a HLA–B514. La prevalencia más alta se ha reportado en Turquía (420/100,000 habitantes) y en Estambul (380/100,000 habitantes). La enfermedad es raramente vista en el oeste, el norte de Europa y EE. UU.. En Alemania se ha reportado una prevalencia de 2.26/100,000, y en EE. UU. de 5.2/100,0003. Aunque hay reportes de enfermedad de Behçet de otras partes del mundo incluyendo Mongolia, Rusia, Brasil, México, Colombia, Argentina, Chile, Cuba, Australia y Nueva Zelanda, estos reportes acumulan menos de 200 pacientes2. Esta distribución geográfica se ha referido como evidencia que apoya la influencia genética en la patogenia de la enfermedad3. En pacientes mexicanos con enfermedad de Behçet se ha encontrado una frecuencia elevada de HLA-B*44, HLA-B*52, HLA-B*56, HLA-DRB1*01 y HLA-DRB1*134. La baja frecuencia de marcadores nativos en los pacientes mestizos mexicanos con enfermedad de Behçet sugiere que la mezcla genética entre Medio Oriente, orientales y amerindios es un evento que aumentó el riesgo de desarrollar la enfermedad en la población mexicana4.
La anormalidad de la respuesta inmune innata y adaptativa desempeña un papel muy importante en la enfermedad de Behçet5. Algunos estudios han demostrado que HLA-B*51 está asociado a la enfermedad de Behçet con su positividad en más del 60% de los pacientes. Además, algunos metanálisis han identificado IL-10, IL-23R, IL-12B2 fuertemente asociados a la enfermedad de Behçet. La IL-23 es una citocina proinflamatoria que estimula la proliferación de Th17, aumenta la producción de citocinas inflamatorias y aumenta la expresión de IL-23 p19 mRNA en las lesiones dermatológicas tipo eritema nudoso encontradas en pacientes con enfermedad de Behçet activa2. Muchos estudios han revelado el dominio de las células Th1 en la enfermedad y estudios recientes han sugerido el involucramiento de células Th17 en su patogenia. Además, varias investigaciones han reportado que las células T reguladoras están paradójicamente aumentadas en la enfermedad. Se ha establecido el rol de la hipersensibilidad dominada por Th1 debido a la pérdida del equilibrio Th1/Th26.
Es posible que las proteínas de choque térmico regulen la diferenciación de células T mediante varias interacciones. Las interacciones entre el receptor de linfocitos T y péptidos derivados de proteínas de choque térmico con el complejo mayor de histocompatibilidad y entre IL-12 y su receptor podrían inducir una diferenciación de células T aberrante en pacientes con enfermedad de Behçet7.
Las células Th17 producen un número de citocinas proinflamatorias como IL-17A, IL-17F, IL-21 e IL-22, mientras que el TGF-beta y la IL-6 son esenciales para su desarrollo6. Hay evidencia de que estas células son patológicas en muchas enfermedades autoinmunes e inflamatorias. La información de cómo las células Th17 interactúan con otras células inmunes es limitada, mas sin embargo datos recientes sugieren que las células Th17 no son reguladas estrictamente por las células T reguladoras5.
Las reacciones autoinmunes o autoinflamatorias en la enfermedad de Behçet atacan primeramente los vasos sanguíneos2. El endotelio es el objetivo principal en la patogenia8. Existe confusión sobre la tendencia trombótica en la enfermedad, en términos de si la hipercoagulabilidad encontrada es primaria o es secundaria a la inflamación9. Mediante una producción de autoanticuerpos anticélulas endoteliales, se desarrolla una vasculitis y el consiguiente daño tisular1. Se ha identificado la alfa enolasa como un antígeno blanco de anticuerpos antiendoteliales tipo IgM en pacientes con enfermedad de Behçet. Los anticuerpos antiendoteliales se unen a las células endoteliales, dando como resultado activación celular con la secreción aumentada de quimioatrayentes y/o citocinas y la secreción o inhibición de prostaciclina2.
Existen criterios diagnósticos establecidos por el Grupo de Estudio Internacional de la Enfermedad de Behçet (tabla 1)1. No hay un examen de laboratorio específico para la enfermedad10. La enfermedad se manifiesta por la aparición de úlceras recurrentes en boca y genitales y lesiones oculares y dermatológicas. Las lesiones articulares también son frecuentes, además de la afección al sistema nervioso y vascular11. Entre las manifestaciones oculares se encuentran la uveítis anterior, con o sin hipopion, infiltración celular y opacificación vítrea, vasculitis retiniana, infiltrados y microhemorragias retinianas, oclusiones de vena central de la retina, edema macular cistoide e hiperemia papilar1. Afortunadamente la mayoría de las manifestaciones son autolimitadas con excepción de las oculares, que pueden causar ceguera11. La enfermedad de Behçet puede ser el mejor ejemplo de un padecimiento caracterizado principalmente por el involucramiento de la vasculatura retiniana que frecuentemente tiene resultados devastadores en la visión12.
Criterios diagnósticos del Grupo de Estudio Internacional de la Enfermedad de Behçet
| Úlceras orales recurrentes |
| Lesiones aftosas mayores o menores o lesiones herpetiformes observadas al menos 3 veces dentro de un periodo de 12 meses |
| Presencia de 2 criterios siguientes |
| Ulceración genital recurrente: Ulceración aftosa o cicatriz |
| Lesiones oftalmológicas: Uveítis anterior y/o posterior, celularidad vítrea, vasculitis retiniana |
| Lesiones dermatológicas: Eritema nudoso, seudofoliculitis, lesiones papulopustulares, nódulo acneiforme en pacientes postadolescentes (que no se encuentren bajo tratamiento esteroideo) |
| Prueba de patergia positiva: Leído por un médico entre las 24-48h |
Las metas de la terapia en la enfermedad de Behçet son suprimir la inflamación, reducir la frecuencia y severidad de recurrencias y minimizar el involucramiento de la retina. Para ser efectivo, el tratamiento debe ser implementado tempranamente. En casos de uveítis leve como uveítis anterior, corticoesteroides tópicos junto con midriáticos y ciclopléjicos pueden controlar la enfermedad. El siguiente paso deben ser corticoesteroides sistémicos. Estos son utilizados en ataques inflamatorios oculares agudos, panuveítis y vasculitis de retina13.
Los corticoesteroides suprimen efectivamente todas las fases de involucramiento ocular en la enfermedad de Behçet. Aunque estos fármacos no previenen el deterioro visual, los corticoesteroides sistémicos son efectivos especialmente al ser utilizados en conjunto con otros agentes inmunosupresores14.
El clorambucilo fue el primer agente citotóxico utilizado en el tratamiento de enfermedad de Behçet ocular. Este fármaco alquilante de lenta acción puede ser administrado en casos ambulatorios. La ciclofosfamida es un agente alquilante superior a los corticoesteroides en el control de la inflamación en la enfermedad de Behçet pero su toxicidad a la médula ósea limita su uso. Como la ciclofosfamida actúa más rápido y es más tóxica que el clorambucilo, su uso se reserva para casos muy refractarios. La azatioprina es un derivado de la mercaptopurina que es efectivo en el tratamiento de la enfermedad de Behçet14. Este fármaco es de elección, junto con la ciclosporina, en casos en donde no hay una respuesta con el manejo con corticoesteroides y es necesario un tratamiento más agresivo13.
La ciclosporina inhibe la activación de linfocitos T y consecuentemente es más segura que los agentes citotóxicos, a pesar de esto, pueden ocurrir complicaciones renales. Se ha observado un efecto rebote al suspender la terapia con ciclosporina. Estos factores han limitado su uso para el tratamiento de la enfermedad de Behçet; aun así, en un estudio realizado en Japón se encontró que la dosis inicial limitó la frecuencia de ataques oculares inflamatorios en un 70% de los pacientes que tenían previamente enfermedad refractaria14.
El tacrolimus es un fármaco inmunosupresor con actividad inmunológica similar a la ciclosporina. Se une a la glucoproteína ácida alfa 1 en suero y selectivamente inhibe linfocitos T CD4+. Japón ha reportado resultados favorables en uveítis refractaria con enfermedad de Behçet14.
La colchicina es un alcaloide que interfiere con la función microtubular que resulta en disfunción de neutrófilos; controla la enfermedad de Behçet aunada al uso de menores dosis de inmunosupresores. En Japón es considerado el fármaco de elección por sus escasos efectos adversos14.
El rituximab es un anticuerpo monoclonal contra CD20. Davatchi et al. realizaron un estudio en donde se observó su eficacia en manifestaciones oculares severas15. Asimismo se ha reportado mejoría de lesiones mucocutáneas severas, así como del involucramiento neurológico y ocular con el uso de metotrexate13. También se encontró que el mofetil micofenolato es seguro y efectivo para controlar el edema macular y en la reducción de las recurrencias de uveítis en pacientes que no responden al tratamiento inmunosupresor tradicional. Estos fármacos corresponden a la tercera línea de tratamiento13.
Aparentemente los mejores resultados son obtenidos mediante la combinación de fármacos como los corticoesteroides con agentes citotóxicos o ciclosporina. A pesar de esto no existe un tratamiento concluyente y seguro para los pacientes con enfermedad de Behçet2.
Los interferones son una gran familia de glucoproteínas con propiedades antivirales, antitumorales e inmunomoduladoras. Se utilizan como tratamiento de la enfermedad por la asociación que existe con infecciones virales y por sus efectos biológicos como la habilidad para mejorar la actividad de las células natural killer e inhibir las células T gamma delta. Los mejores resultados se han visto en manifestaciones oculares severas y/o refractarias. Los efectos adversos son frecuentes, dosis dependientes, pero no son severos14.
El uso de bloqueadores del factor de necrosis tumoral en la enfermedad de Behçet está dado por la fuerte implicación del factor de necrosis tumoral alfa en la patogénesis de la enfermedad. Los pacientes con enfermedad activa demuestran un numero de monocitos y linfocitos T aumentados expresando receptores gamma delta que sobreproducen factor de necrosis tumoral alfa14. El infliximab, etanercept y adalimumab han tenido éxito en pacientes con enfermedad de Behçet que tienen además lesiones mucocutáneas o gastrointestinales, neurológicas o aneurismas pulmonares. Hay suficientes publicaciones que indican un avance terapéutico para pacientes que son resistentes a regímenes inmunosupresores estándar y para pacientes con contraindicaciones o intolerancia al tratamiento2.
Se están llevando a cabo investigaciones para revelar la patogénesis y desarrollar nuevos agentes terapéuticos. La tolerancia inmune utilizando los péptidos de proteínas de choque térmico 65/60 ha sido recientemente considerada como una posible terapia para pacientes con enfermedad de Behçet2. Además, el trasplante autógeno de células madre hematopoyéticas ha demostrado eficacia en algunos casos14.
El manejo más efectivo involucra un diagnóstico temprano y la intervención clínica con seguimiento continuo. Como consecuencia será posible reducir el riesgo de complicaciones serias y costos socioeconómicos debidos a la enfermedad de Behçet2.
A continuación se presentan 2 casos clínicos, su evolución y su seguimiento.
Presentación de casosSe trata de un paciente femenino de 33 años de edad que ingresa al servicio de oftalmología por presentar hiperemia conjuntival y disminución de agudeza visual de ojo izquierdo de 5 meses de evolución.
A la exploración oftalmológica, presenta una agudeza visual de ojo derecho de 20/30 (que mejora a) 20/20 (−1) y ojo izquierdo de PMM que no mejora. La presión intraocular de ambos ojos fue de 15mmhg.
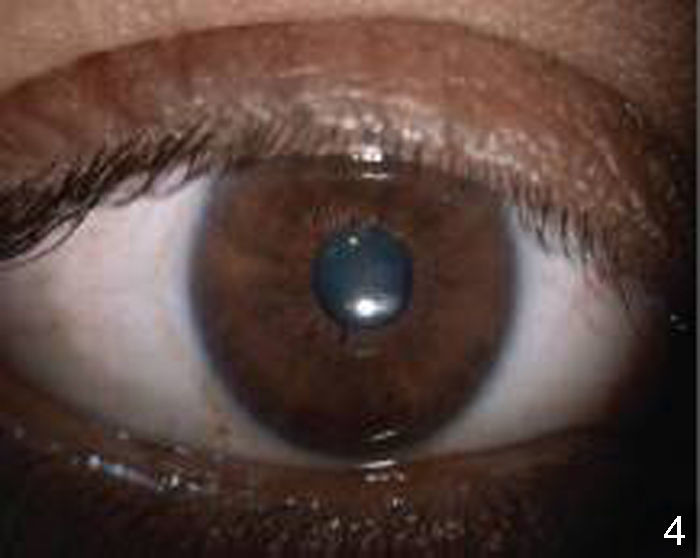
Los anexos de ambos ojos y el segmento anterior del ojo derecho se encontraban sin alteraciones (fig. 1). En el ojo izquierdo se observa una conjuntiva normocrómica, fondo de saco libre, córnea con depósitos retroqueráticos finos, cámara anterior formada, con celularidad +1, iris con crestas y valles, sinequias posteriores de MIII a MIX, pupila hiporrefléctica, pigmento en cápsula anterior de cristalino y cristalino transparente (fig. 2).


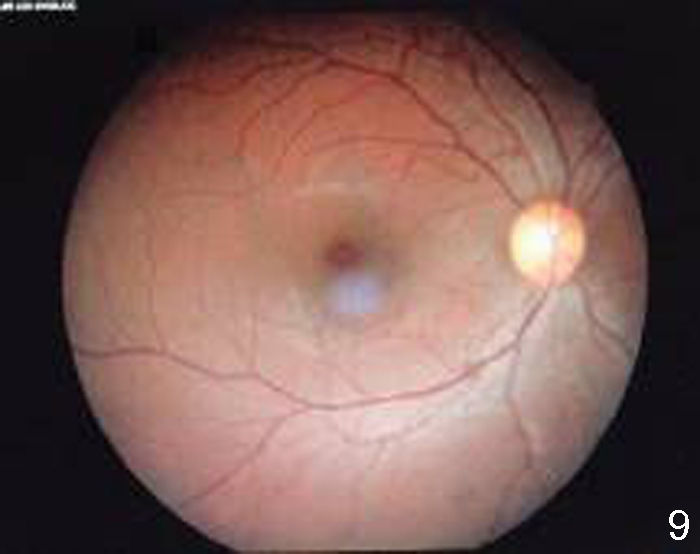
El fondo de ojo del ojo derecho se observó sin datos patológicos (fig. 3). En el ojo izquierdo los medios turbios a expensas de vitreítis 2+ permitieron valorar una papila hiperémica con bordes ligeramente borrados y emergencia central de vasos (fig. 4).


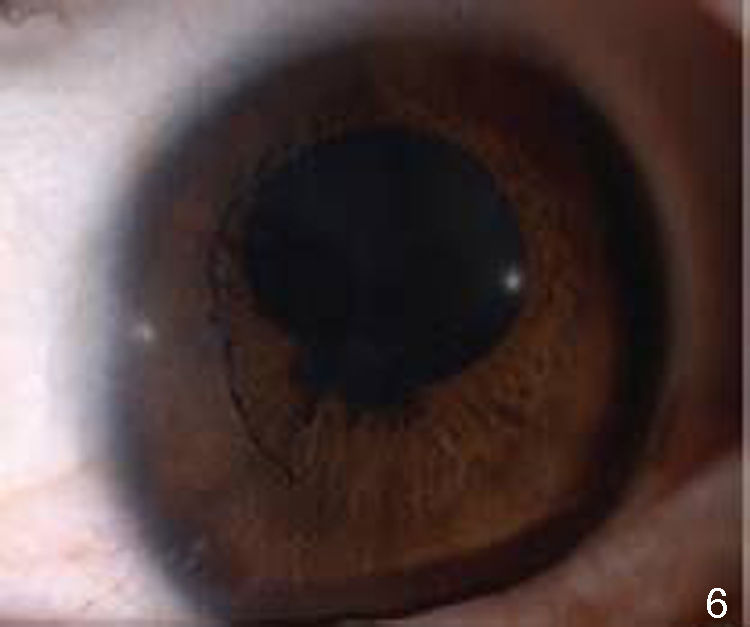
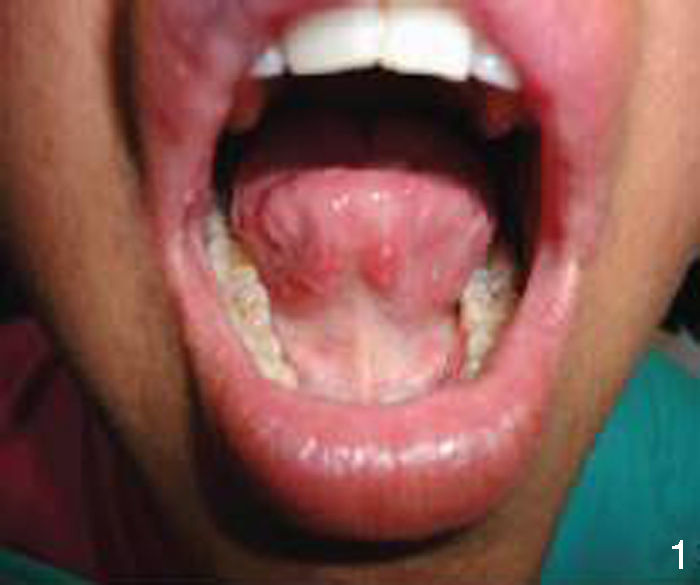
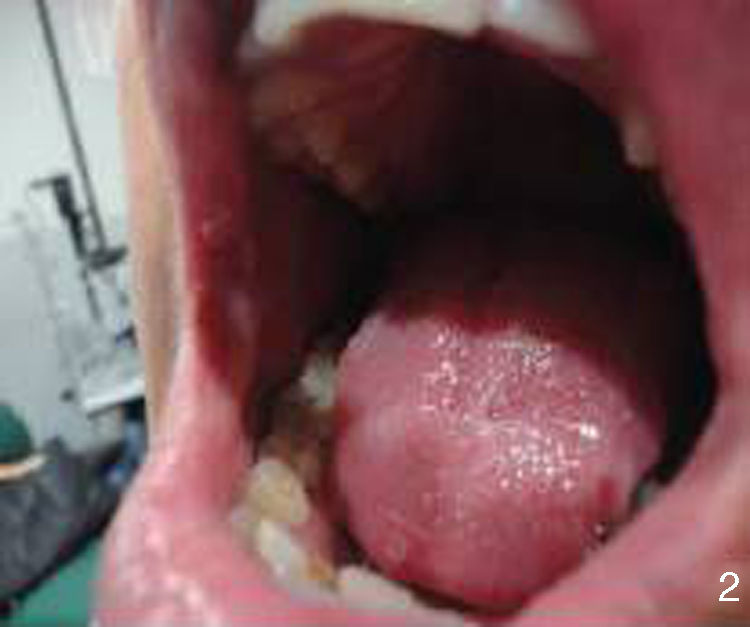
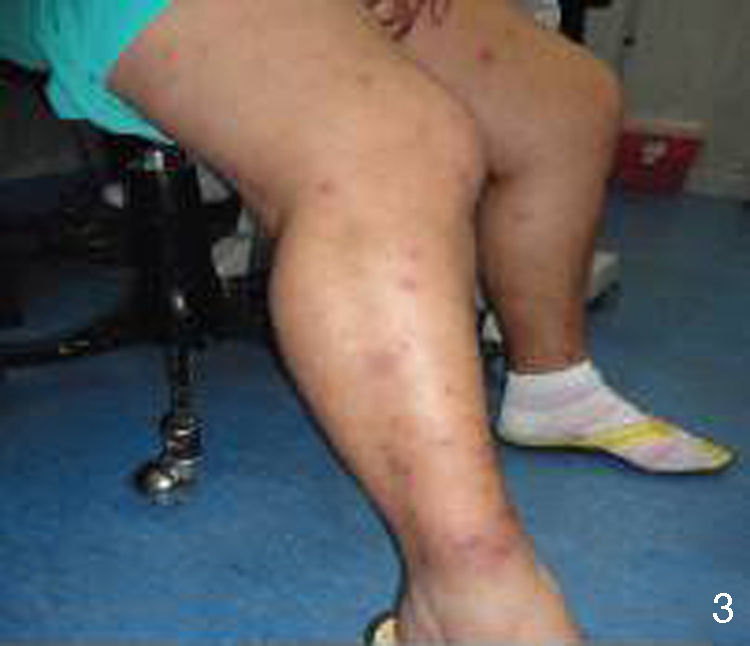
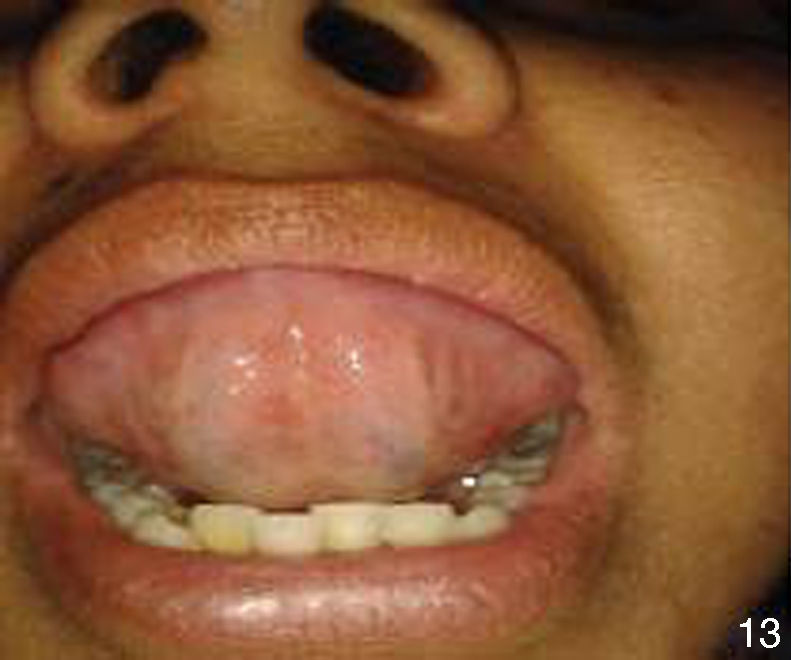
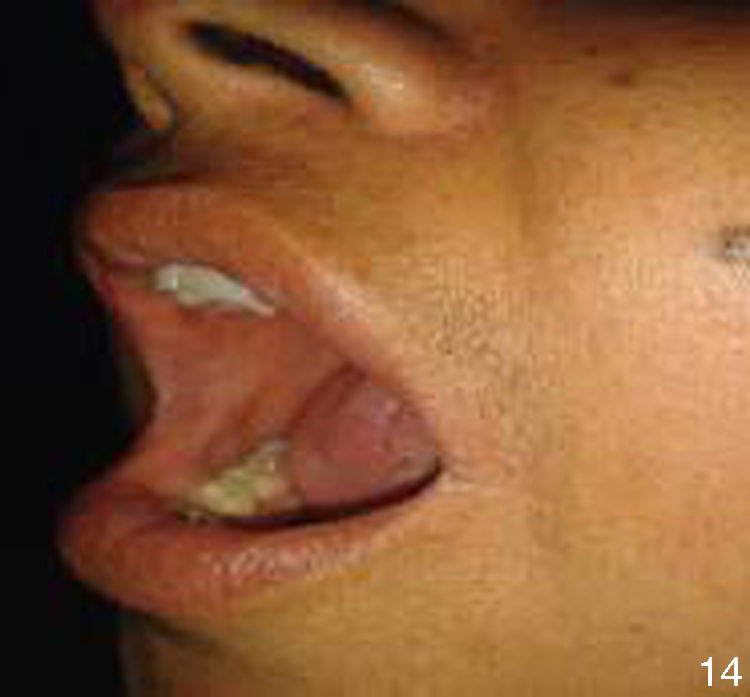
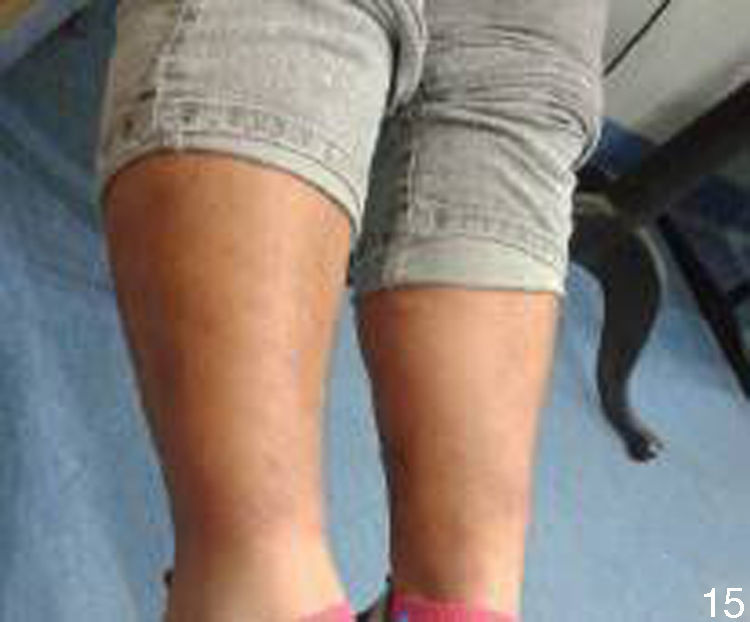
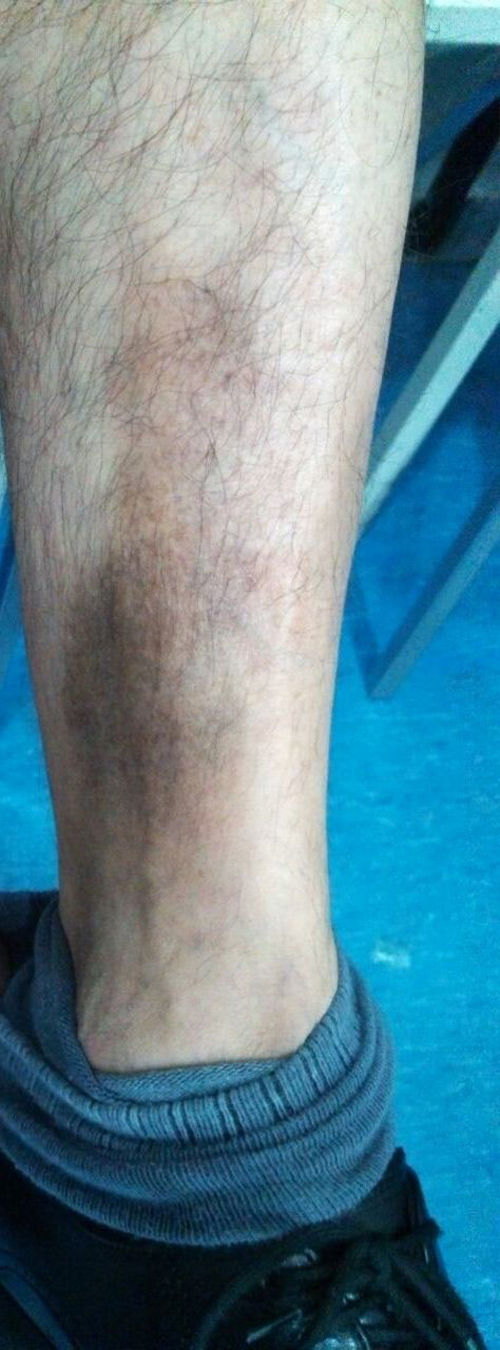
Al interrogatorio la paciente refiere varios episodios de hiperemia ocular izquierda y aftas orales con remisión espontánea, desde hace aproximadamente un año. Con este antecedente se realiza la exploración física, observando úlceras en lengua y carrillo derecho (figs. 5 y 6) y miembros inferiores con lesiones violáceas de borde irregular, con induración a la palpación, no dolorosas (fig. 7).



Se realizaron pruebas de laboratorio con biometría hemática, química sanguínea y examen general de orina dentro de la normalidad; FTA-ABS, VDRL y PPD negativos.
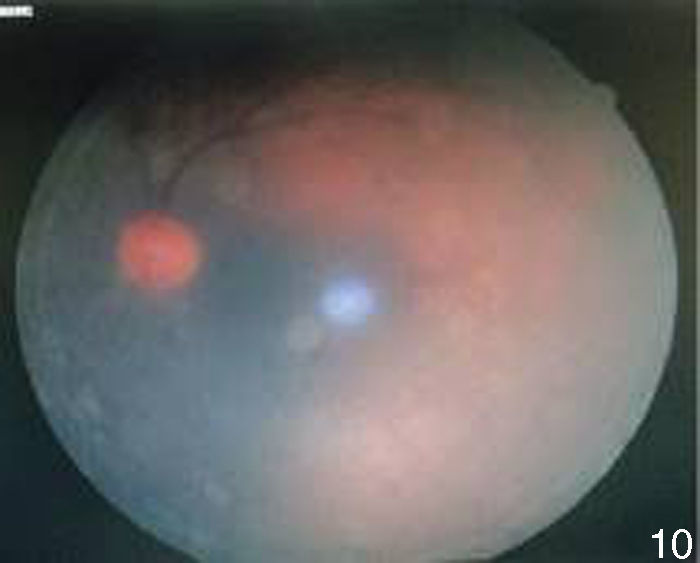
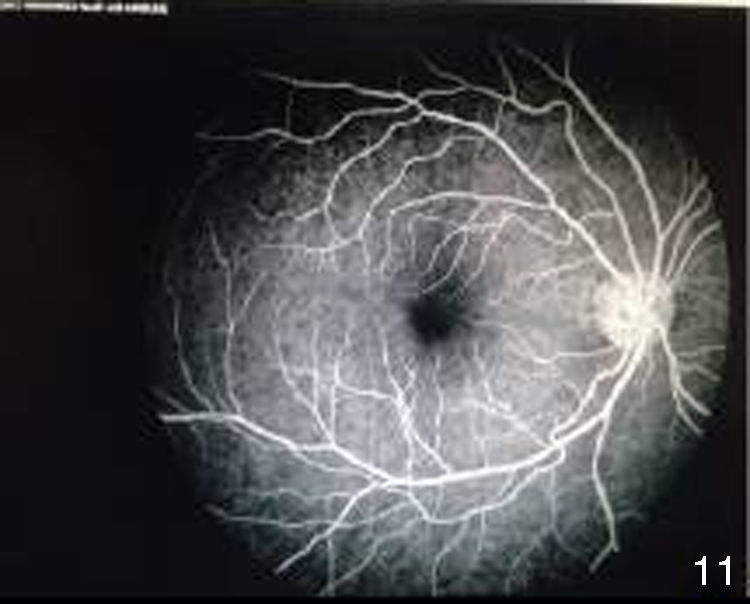
Se realizó fluorangiografía, que evidenció el ojo derecho sin alteraciones (fig. 8) y el ojo izquierdo con zonas de hiperfluorescencia por fuga en fase arteriovenosa tardía (fig. 9).


Posteriormente, en conjunto con el servicio de Dermatología se decide realizar biopsia de úlcera oral reportando vasculitis leucocitoclástica, abundante infiltrado inflamatorio de tipo linfohistiocitario, con neutrófilos y zonas con destrucción parcial de los vasos capilares. Asimismo se realizó prueba de patergia, la cual resultó positiva.
Ante estos resultados y la sospecha clínica (uveítis, úlceras orales, eritema nudoso) se confirma el diagnóstico de enfermedad de Behçet y se comenta al servicio de Reumatología con quien se maneja en conjunto a base de azatioprina 75mg vo/24h, prednisona 50mg vo/24h, prednisolona una gota/4h, y tropicamida fenilefrina una gota/8h.
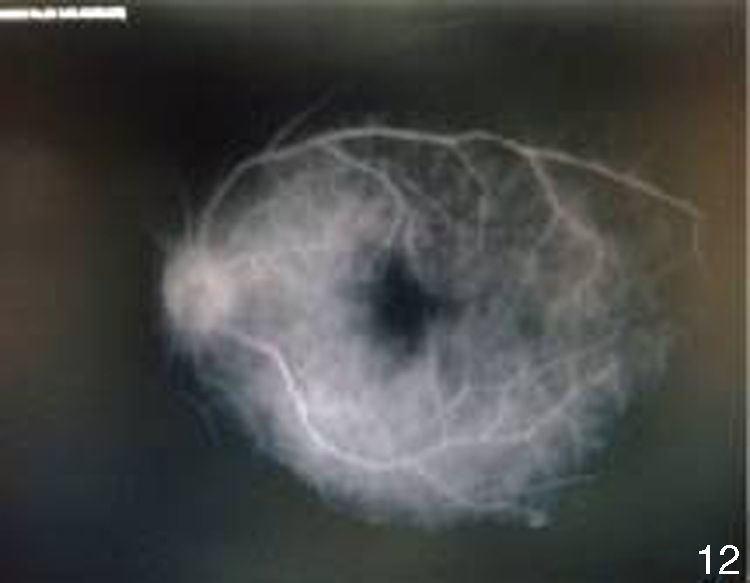
A 3 meses del tratamiento la agudeza visual de ojo izquierdo mejoró a cuenta dedos a 2m. Las úlceras orales remitieron totalmente y el eritema nudoso de manera parcial (figs. 10–12). El segmento anterior del ojo izquierdo continuaba con sinequias posteriores, de MIII–MIX y pigmento en cápsula anterior de iris (fig. 13). En el fondo de ojo se observaron medios claros que permitieron valorar palidez de nervio óptico, pliegues en área macular y zonas de envainamiento vascular periféricas. La fluorangiografía en fase arteriovenosa tardía presentó hiperfluorescencia por fuga (figs. 14 y 15).






Debido a la disminución de la agudeza visual notable y no recuperable, además de los hallazgos a la exploración se le realizo OCT macular que fue normal (fig. 16).

El segundo caso es el de un paciente masculino de 49 años de edad, que acude al servicio de Urgencias de Oftalmología por referir hiperemia conjuntival y dolor ocular izquierdo de una semana de evolución. Entre sus antecedentes patológicos, tiene diagnóstico de enfermedad de Behçet realizado hace un año, actualmente en tratamiento por Reumatología con azatioprina 50mg una tableta/12h. El paciente niega antecedentes oftalmológicos.
A la exploración oftalmológica, presenta agudeza visual de ojo derecho de 20/20 y de ojo izquierdo de 20/25. Presión intraocular de ambos ojos de 12mmhg.
Anexos oculares sin alteraciones. Segmento anterior de ojo derecho sin alteraciones. Ojo izquierdo con conjuntiva hipéremica difusa leve, córnea transparente, cámara anterior formada con celularidad de 0.5+, iris con crestas y valles, pupila refléctica y cristalino transparente.
El fondo de ojo derecho se encontraba sin datos patológicos. El fondo de ojo izquierdo constaba de medios claros, papila fisiológica, brillo foveolar presente, cicatriz coriorretiniana inferior y retina aplicada.
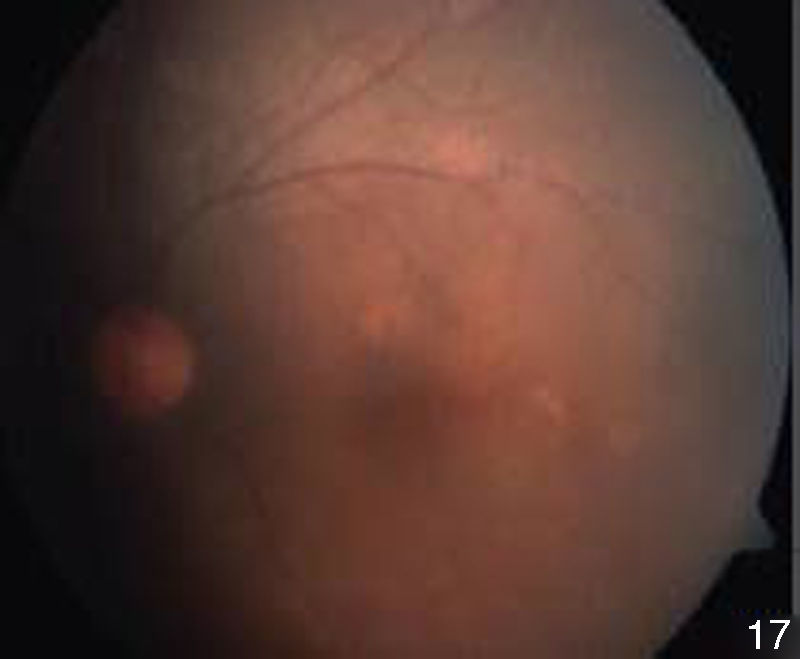
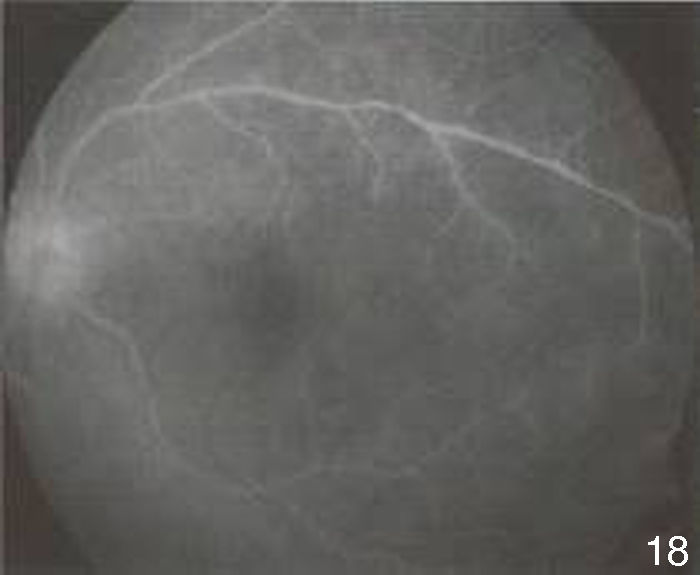
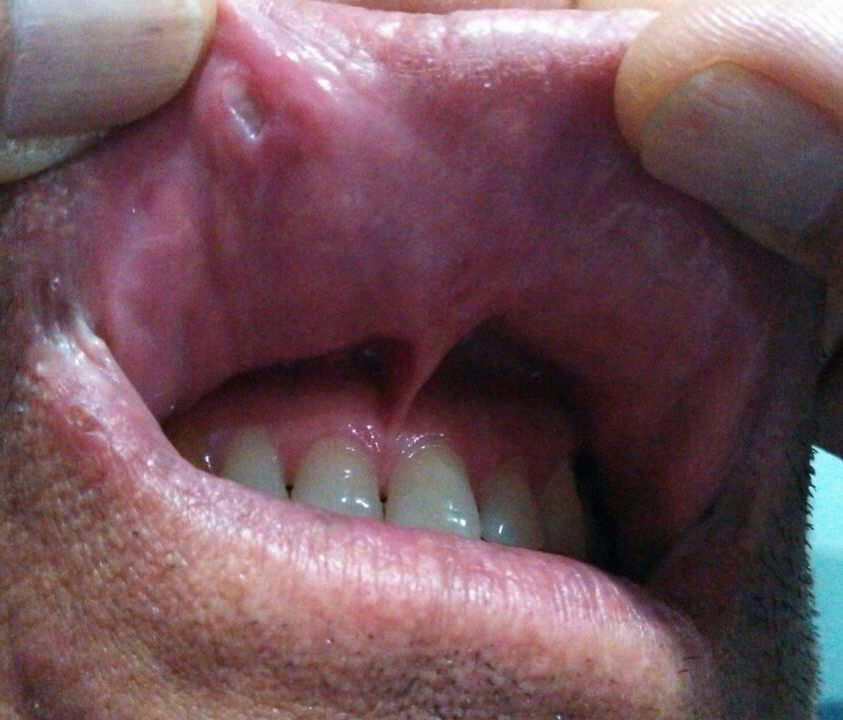
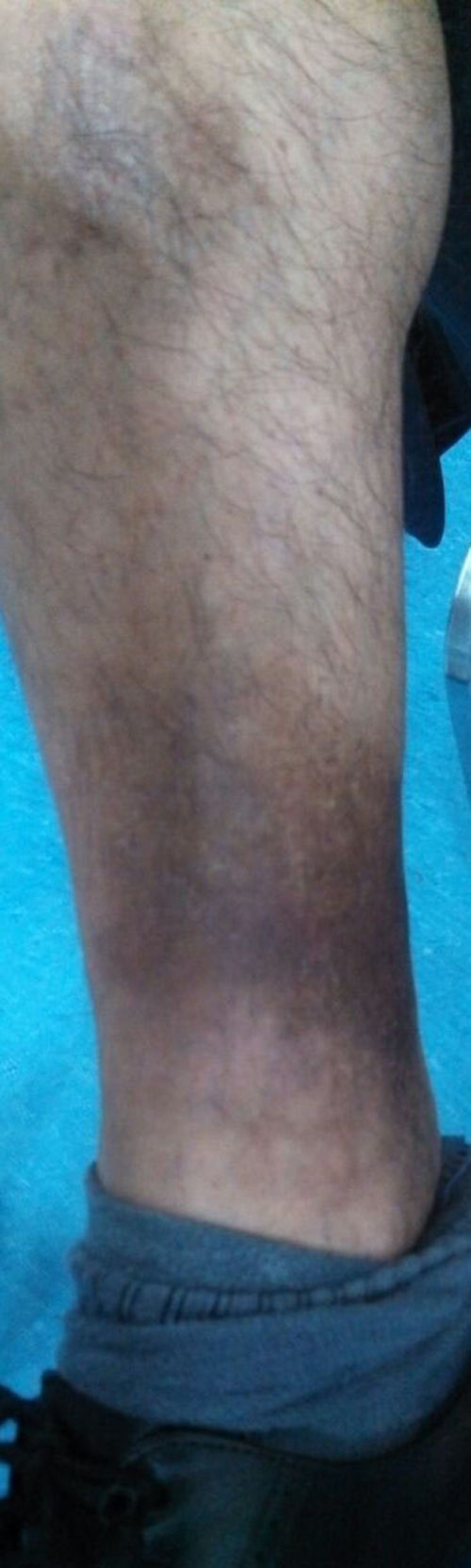
A la exploración física se observan úlceras en mucosa bucal (fig. 17) y lesiones violáceas en forma de placas descamativas induradas, no dolorosas, en miembros inferiores (figs. 18 y 19).



Se inició tratamiento a base de prednisolona gotas oftálmicas a dosis; reducción con mejoría sintomatológica y clínica.
DiscusiónLas manifestaciones oculares de la enfermedad de Behçet se presentan inicialmente de manera bilateral en el 80% de los pacientes y aproximadamente aparecen 3 a 4 años posteriores a las manifestaciones cutáneas y bucales1. Por otro lado, las manifestaciones iniciales pueden ser unilaterales, pero progresan de manera bilateral en dos tercios de los casos. La inflamación no granulomatosa con vasculitis obliterativa necrosante se puede observar tanto en segmento anterior como posterior, o más comúnmente, en ambos8. Son pocos los casos presentados en la literatura sobre manifestaciones oculares en enfermedad de Behçet en México. Tenorio y Velázquez reportan un caso clínico con inicio de tratamiento sistémico posterior a 10 años de evolución de la enfermedad de Behçet, con afección visual bilateral y recurrente a pesar del tratamiento con talidomida y prednisona, lo que generó su cambio a prednisona, azatioprina y metotrexato, quedando así en vigilancia16. En nuestro primer caso la paciente se presenta con panuveítis unilateral; debido al cuadro clínico se confirmó enfermedad de Behçet y se indicó tratamiento tópico y sistémico. El manejo se realizó en conjunto con Reumatología para su seguimiento. Se observó mejoría clínica con prednisona y azatioprina pero la mejora en agudeza visual fue mínima en el ojo izquierdo. El inicio del tratamiento tuvo lugar un año después del comienzo de la sintomatología sistémica.
Hay falta de evidencia en cuanto al tratamiento de la enfermedad de Behçet y en la afectación sistémica se recomienda el uso de inmunosupresores, siendo la azatioprina aceptada como de primera elección en estos casos17. La paciente de nuestro caso se ha mantenido en vigilancia y la baja visual presentada nos lleva a pensar en la probable atrofia óptica que se presenta en al menos una cuarta parte de los pacientes con enfermedad de Behçet; la atrofia óptica progresiva puede ocurrir como resultado de microvasculitis de las arteriolas que irrigan el nervio óptico y la inflamación ocular repetitiva1.
Tenorio Velázquez también presentan un segundo caso clínico en donde se menciona la presentación de la enfermedad como panuveítis unilateral con hipopion y vasculitis de retina para lo cual se inicia tratamiento tópico con prednisolona y atropina con posterior interconsulta al servicio de Reumatología16. Es de gran importancia el tratamiento multidisciplinario que requieren los pacientes con enfermedad de Behçet y la atención debe de ser prioritaria. Como muestra nuestro segundo caso, el paciente acude al servicio con una inflamación ocular unilateral mínima, lo que es una presentación rara de la enfermedad. Sin embargo, el paciente ya se encontraba con diagnóstico de enfermedad de Behçet y en tratamiento sistémico con azatioprina, lo cual puede ser la razón de la mínima afección oftalmológica la cual se pudo controlar con tratamiento tópico a base de prednisolona.
El pronóstico en un paciente con afección de polo posterior es muy malo. Además, las manifestaciones oculares y neurológicas suelen progresar a pesar de tratamiento. El retraso en el diagnóstico, frecuente en los países con baja prevalencia, incrementa la morbimortalidad de los pacientes con esta enfermedad18.
ConclusiónLa enfermedad de Behçet se caracteriza por la recurrencia de la inflamación, la cual involucra tanto el segmento anterior como el posterior y puede afectar la visión de manera importante. Los corticoesteroides se utilizan para controlar la inflamación, pero los inmunosupresores son los medicamentos utilizados para prevenir recurrencias. La terapia biológica ha mostrado eficacia en el manejo de las manifestaciones de la enfermedad, sin embargo, son necesarios estudios con un mayor número de pacientes y un seguimiento prolongado. El pronóstico depende del involucramiento clínico y, a pesar del tratamiento, se tiene un pronóstico visual pobre, llevando a la ceguera a un 25% de los pacientes con enfermedad de Behçet. La investigación de la patogenia y los tratamientos que puedan mejorar la calidad de vida de un paciente con enfermedad Behçet y prevenir sus terribles complicaciones es un tema actual que indiscutiblemente tiene muchos pasos a seguir.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoLas autoras no recibieron patrocinio para llevar a cabo este artículo.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.
